Background
Prior long-term-care (LTC) studies confound anatomic reservoir and pathogen effects when multiple multidrug-resistant organisms (MDROs) circulate. We created a four-tier clearance framework to disentangle site- and species-specific behavior and quantify decolonization, mortality, and asymmetric co-colonization.
Methods
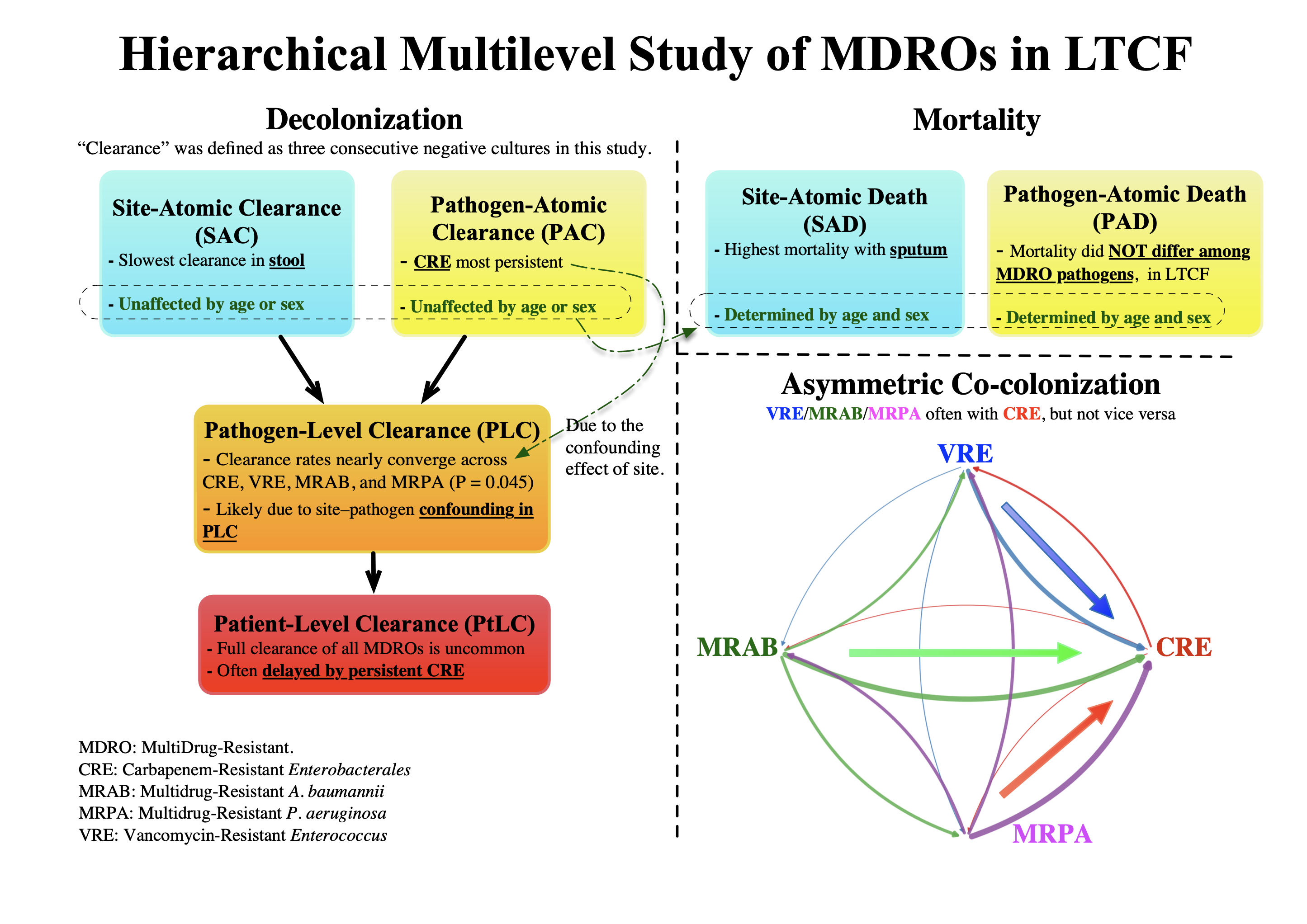
Between January 2024 and May 2025, 98 LTC residents colonized with carbapenem-resistant Enterobacterales (CRE), vancomycin-resistant Enterococcus (VRE), or multidrug-resistant Pseudomonas aeruginosa (MRPA) or Acinetobacter baumannii (MRAB) underwent weekly stool, urine, sputum, wound, blood cultures, yielding a total of 2,772 specimens. Clearance—operationally defined as three consecutive negatives—was analyzed across four tiers (site-atomic, pathogen-atomic, pathogen-level, patient-level). Conditional-probability tables were constructed to summarize how frequently the four MDROs co-colonized the same patient.
Results
Clearance was slowest in stool and for CRE, independent of patient attributes of age or sex. In this LTCF cohort, mortality depended on sputum carriage and host factors rather than pathogen identity. More than half of residents carried multiple MDROs, and conditional-probability analysis revealed asymmetric co-colonization: non-CRE organisms almost always co-colonized with CRE, whereas the reverse was uncommon.
Conclusions
The hierarchical analysis showed that, contrary to common expectations, decolonization was independent of patient attributes (age and sex) and that, in this LTCF setting, mortality was unrelated to pathogen identity. Asymmetric co-colonization therefore warrants automatic CRE screening following MRAB, MRPA, or VRE isolation, and underscores the need to investigate directional co-colonization patterns among multiple MDRO co-colonizations commonly observed in LTCFs—as exemplified by vanA transfer from VRE to MRSA (methicillin-resistant Staphylococcus aureus) yielding VRSA (vancomycin-resistant Staphylococcus aureus).
Introduction
Multidrug-resistant organisms (MDRO) infections pose a critical global health threat and are associated with worse outcomes than drug-susceptible infections [1]. Although most MDRO research has focused on acute-care or tertiary hospital settings, long-term care facilities (LTCF) are increasingly recognized as significant reservoirs of resistant pathogens [2]. Outcomes of MDRO infections vary by infection site, organism type, and the presence of polymicrobial infections [3, 4]. Co-colonization—where patients harbor multiple MDROs—is being reported frequently in acute and post-acute settings, further complicating infection control and worsening clinical outcomes [5].
Prior longitudinal studies of MDRO carriage exhibit one or more of the following limitations: single‐pathogen focus, retrospective design, limited sample sizes, short‐term study periods, heterogeneous follow-up intervals, and complex symptom-driven follow-up endpoints [6-11]. More critically, they seldom account for the intertwined effects of anatomical site and pathogen, and almost never examine the directional, asymmetric co‐colonization of multiple MDROs—leaving two critical gaps unaddressed. First, intra‐patient heterogeneity is obscured: some pathogens decolonize rapidly while others prolong quarantine [11], and different anatomical sites (e.g., sputum versus stool) exhibit markedly divergent decolonization kinetics for the same pathogen [12]. Second, the asymmetric nature of co‐colonization is overlooked: the presence of one pathogen often predicts carriage of another, yet the reverse association may be weak [13].
In the absence of a routinely recommended decolonization strategy for MDRO carriage, understanding clearance dynamics is critical, as infection-control policies in LTCFs must rely on natural clearance. Building on prior work and within existing clinical constraints, a prospective cohort study was conducted in a Korean LTCF, tracking a sizable resident population over an extended period during which hundreds of adjudicated events—clearance (operationally defined here as three consecutive negative cultures), death, or censoring—were captured and classified within a four-tier analytic hierarchy to minimize confounding by anatomical reservoir and pathogen. This four-tier framework, combined with conditional-probability analysis, provides a rigorous method for quantifying decolonization dynamics, mortality, and the directional co-colonization patterns that emerge when multiple MDROs circulate.
Methods
Study design and setting
This prospective cohort study of MDRO colonization dynamics was conducted from January 2024 through May 28, 2025, at Seoul Smart Convalescent Hospital (SSCH), a LTCF in the Republic of Korea. Patients were placed in single rooms whenever possible; if capacity was exceeded, cohort isolation was applied in multi-bed rooms. Adults aged ≥18 years were enrolled if they had colonization at baseline, defined as (i) an MDRO-positive culture detected on admission screening or during symptom-driven testing within the facility, or (ii) transfer from another hospital with documented MDRO carriage. Although SSCH routinely admits patients retrospectively identified as MDRO carriers at other hospitals for quarantine, all surveillance data in this study were collected prospectively after enrollment. “Clearance” was operationally defined, per Korea Disease Control and Prevention Agency (KDCA) guidelines, as three consecutive negative cultures—each obtained at least three days apart—from every anatomic site that had previously yielded a target organism; we used this definition as a clear endpoint for observing quarantined patients. Participants were followed until clearance, death, administrative censoring on May 28, 2025, or censoring at the date of the last weekly culture due to permanent discharge, transfer without return, or withdrawal of consent, whichever occurred first. Patient attributes (age and birth-assigned biological sex) and clinical endpoints, including in-hospital death, were extracted from the electronic medical record. The Institutional Review Board of SSCH—registered with the Korean Ministry of Health and Welfare (Registration No. 3-70094812-AB-N-01)—approved the study protocol (IRB No. 2024-CR-001; approved January 2, 2024), and all participants (or their legal guardians) provided written informed consent in accordance with the Declaration of Helsinki and Korean Good Clinical Practice guidelines.
Eligibility criteria and target organisms
Eligible participants were adult inpatients colonized with at least one of four prespecified MDROs—carbapenem-resistant Enterobacterales (CRE), vancomycin-resistant Enterococcus (VRE), multidrug-resistant Pseudomonas aeruginosa (MRPA), or multidrug-resistant Acinetobacter baumannii (MRAB), where KDCA mandates isolation exclusively for CRE and leaves isolation of other MDROs to institutional discretion. Residents whose only MDRO fell outside these four categories—such as carbapenem-resistant Pseudomonas aeruginosa (CRPA) or carbapenem-resistant Acinetobacter baumannii (CRAB)—were excluded because KDCA guidelines specify MRPA and MRAB, not CRPA or CRAB, for quarantine. Species were identified by MALDI-TOF MS, and antimicrobial susceptibility testing was performed with the VITEK-2 system; interpretations followed CLSI M100 breakpoints [14].
Surveillance culture protocol
Upon first documentation of MDRO carriage, residents were enrolled in a structured surveillance program. At baseline, rectal swabs and urine cultures were obtained to screen for CRE and VRE, and urine and sputum cultures with susceptibility testing were collected for all four target organisms. Thereafter, once an anatomic site tested positive for any target MDRO, we obtained weekly rectal swabs, urine specimens, and sputum specimens from that site. Cultures were collected at least once per week, even if a resident was briefly transferred to a nearby acute-care hospital for treatment or procedure and then readmitted; this ensured at least one culture per week. Missing a weekly culture constituted an exclusion criterion; specifically, patients who could not complete uninterrupted weekly surveillance—such as those briefly transferred and readmitted—were excluded. To allow peers to examine the full culture sequence, all results have been released in Supplementary Dataset S1 (RawDataset.zip) for complete transparency and potential secondary analysis. Rigorous compliance was enforced by national policy: the Korean Health Insurance Review and Assessment Service (HIRA) withholds reimbursement for isolation care if mandated cultures are skipped. Blood and wound specimens were obtained only when clinically indicated or when a referring facility had already reported a positive result. Stool cultures were omitted for MRPA and MRAB, as these non-fermenters rarely colonize the gastrointestinal tract and prior validation studies demonstrated negligible yield [15, 16]. Each isolate was logged by pathogen, anatomic site, and collection date.
The tiers progress from the most granular—site-atomic clearance (SAC) and pathogen-atomic clearance (PAC)—to pathogen-level clearance (PLC) and, ultimately, patient-level clearance (PtLC). Because SAC and PAC are novel terms, detailed plain-language definitions with an illustrative example are provided in Supplementary Note S2. When multiple MDROs are present, with potential conflation between site and pathogen, SAC reflects clearance per individual site, allowing comparisons across the atomic unit of reservoir sites (stool, urine, sputum, wound, blood), whereas PAC reflects clearance with respect to pathogens, allowing comparisons across the atomic unit of the pathogen (CRE, VRE, MRAB, MRPA).
SAC is achieved when a single anatomic site yields three consecutive negative cultures for the given pathogen previously isolated, regardless of culture results from other sites for the same pathogen or from any sites for other pathogens. PAC parallels SAC but shifts the focus to the organism: a PAC event is recorded when a specific pathogen produces three consecutive negative cultures at a given site, independent of its persistence at other sites or the coexistence of other organisms. PLC aggregates all sites within a single patient for one pathogen. It is achieved when every site that ever yielded that pathogen records three consecutive negative cultures, thereby documenting eradication of that specific pathogen from the host. PtLC sits atop the hierarchy and serves as the operational trigger for discontinuing transmission-based precautions. It requires three consecutive negative cultures from every site that has ever harbored any target MDRO in that patient. Although PLC and PtLC might seem similar, they serve distinct purposes: PLC defines eradication of a particular organism from all previously positive sites in a single patient, whereas PtLC extends this concept to the global eradication of every colonizing pathogen.
Statistical Analysis
All analyses were performed using the statistical software R, version 4.4.1 (the R Foundation for Statistical Computing, Vienna, Austria) on macOS. The primary endpoint was time to clearance, measured in days from the index positive culture to the first of three consecutive negative cultures meeting the study’s clearance criteria. The secondary endpoint was all-cause, in-hospital mortality, calculated from the date of the first post-admission culture. Mortality was evaluated only at the atomic strata—site-atomic death (SAD) script and pathogen-atomic death (PAD).
Kaplan–Meier curves were generated for each hierarchical tier to characterize colonization persistence and overall survival; medians and 95 % confidence intervals (CIs) were read directly from the curves [17]. When fewer than 50 % of subjects in a stratum experienced the event, the median was reported as “not reached” (NR); when a Greenwood‐based bound could not be estimated, it was reported as “not estimable” (NE). The effects of age (continuous) and sex on clearance and mortality were assessed with Cox proportional-hazards models, and results are presented as hazard ratios (HRs) with 95 % CIs. Two-sided P values < 0.05 were considered statistically significant.
Pathogen co-colonization was examined by converting the patient-by-pathogen presence–absence matrix into a conditional-probability matrix, P(b | a), representing the probability of isolating pathogen b at any time—preceding, concurrent with, or following—the isolation of pathogen a in the same patient.
Results
Study Population and Culture Episodes
From January 2024 through May 28, 2025, the study enrolled 124 patients (Fig. 1). Twenty-six patients were excluded according to the predefined criteria described in Methods. As a result, 98 patients were included in the analysis. The majority (n = 91, 92.9 %) were transferred from a nearby hospital for isolation, while seven were identified as MDRO carriers at SSCH itself. The median age was 79.5 years (IQR, 69.0–84.0), and 50 (51.0 %) were male. Across this cohort, a total of 2,772 culture episodes were recorded: 1,776 for CRE, 632 for VRE, 133 for MRAB, and 181 for MRPA.
Site-atomic clearance (SAC)
Ninety‐eight LTCF residents generated 274 SAC episodes: stool (n = 115), urine (n = 84), sputum (n = 63), wound (n = 11), and blood (n = 2) (Table 1). Blood is shown for completeness but was excluded from modeling because no clearances or deaths occurred. Kaplan–Meier curves differed significantly across sites (Fig. 2a; log‐rank P < 0.001). Stool was the most persistent reservoir (median time‐to‐clearance, 181d; 95% CI, 134–273), whereas sputum, urine, and wound colonization cleared in median 78d (95% CI, 44–112), 47d (95% CI, 36–82), and 28d (95% CI, 21–NE [upper bound not estimable]), respectively. In a multivariable Cox model using stool as the reference (Table 2), the adjusted clearance rate ratio increased 2.66-fold for sputum (hazard ratio [HR], 2.66; 95% CI, 1.60–4.44), 2.84-fold for urine (HR, 2.84; 95% CI, 1.83–4.40), and 5.25-fold for wounds (HR, 5.25; 95% CI, 2.50–10.99); neither age (HR, 0.999; 95% CI, 0.984–1.014) nor sex (male HR, 1.16; 95% CI, 0.80–1.67) was independently associated with clearance. At the SAC tier, stool exhibited the slowest clearance with marked variation across sites, and clearance was independent of patient age and sex.
Site-atomic death (SAD)
During the same interval, 88 of the 274 site-atomic episodes ended in death (Fig. 2b). Median survival differed significantly by anatomical site (log-rank P = 0.02): it was shortest for sputum-colonized residents (median, 68d; 95% CI, 51–NE), intermediate for stool (144d; 95% CI, 93–244), and longest for urine (189d; 95% CI, 155–NE); wound and blood episodes were too sparse for reliable estimates. In the fully adjusted Cox model using sputum as the reference (Table 2), stool carriage was associated with a 41% lower mortality hazard (HR, 0.59; 95% CI, 0.36–0.97) and urine carriage with a 51% lower hazard (HR, 0.49; 95% CI, 0.26–0.92); the wound–sputum contrast did not reach statistical significance (HR, 0.42; 95% CI, 0.05–3.14). Each additional year of age increased the risk of death by 4.4% (HR, 1.044; 95% CI, 1.022–1.067), and male sex conferred a 90% excess hazard (HR, 1.91; 95% CI, 1.23–2.96). At the SAD tier, mortality was highest in sputum-colonized residents, varied significantly by site, and—unlike clearance—was influenced by age and sex.
Pathogen-atomic clearance (PAC)
All 274 PAC episodes—CRE (n = 156), VRE (n = 68), MRPA (n = 29), and MRAB (n = 21)—were evaluable (Table 1). Kaplan–Meier curves separated sharply by pathogen (Fig. 2c; log-rank P ≈ 1 × 10⁻⁹). CRE was the most tenacious organism, with a median time-to-clearance of 159d (95% CI, 111–246) and a crude clearance rate of 32.1%. In contrast, MRAB cleared in a median 28d (95% CI, 26–NE) with a 76.2% clearance rate; MRPA cleared in 27d (95% CI, 24–94) with a 69.0% rate; and VRE cleared in 61d (95% CI, 51–139) with a 51.5% rate. In a multivariable Cox model using CRE as the reference (Table 2), the adjusted clearance rate ratio was 5.30-fold higher for MRAB (HR, 5.30; 95% CI, 2.96–9.51), 5.12-fold higher for MRPA (HR, 5.12; 95% CI, 3.00–8.75), and 2.04-fold higher for VRE (HR, 2.04; 95% CI, 1.32–3.16); neither age (HR, 0.9965; 95% CI, 0.981–1.012) nor male sex (HR, 1.09; 95% CI, 0.76–1.58) was significant. At the PAC tier, CRE cleared most slowly, VRE intermediately, and MRAB/MRPA cleared fastest; as at the SAC tier, clearance was independent of patient age and sex.
Pathogen-atomic death (PAD)
Of the 274 pathogen-atomic episodes, 88 ended in death (Fig. 2d; log-rank P = 0.80), and the survival curves for CRE, VRE, MRAB, and MRPA were virtually identical. Median survival was 136d (95% CI, 102–189) for CRE and 293d (95% CI, 89–NE) for VRE; medians for MRAB and MRPA were not reached (lower 95% CI bound NE for MRAB and 41 d for MRPA). In a fully adjusted Cox model (Table 2), none of the non-CRE pathogens—MRAB (HR, 0.98; 95% CI, 0.30–3.20), MRPA (HR, 0.90; 95% CI, 0.36–2.27), or VRE (HR, 0.82; 95% CI, 0.48–1.41)—significantly affected mortality risk compared with CRE. Instead, host factors predominated: each additional year of age increased the mortality hazard by 4.8% (HR, 1.048; 95% CI, 1.025–1.071), and male sex conferred a 92% excess hazard (HR, 1.92; 95% CI, 1.24–2.98). At the PAD tier, no specific organism uniquely predicted death in our LTCF cohort; instead, mortality was driven by host factors (age and sex). Notably, clearance at the SAC and PAC tiers was independent of those patient attributes.
Pathogen-level clearance (PLC)
The cohort generated 173 PLC episodes—CRE (n = 85), VRE (n = 44), MRPA (n = 23), and MRAB (n = 21)—of which 14/85 (16.5%), 17/44 (38.6%), 16/23 (69.6%), and 14/21 (66.7%) met the clearance definition, respectively (Table 1). Median time-to-clearance was 68 d (95% CI, 53–246) for CRE, 57d (95% CI, 41–139) for VRE, 26d (95% CI, 21–94) for MRPA, and 28d (95% CI, 27–78) for MRAB. Kaplan–Meier curves appeared broadly similar (Fig. 2e), and the global log-rank test indicated modest differences among pathogens (P = 0.0397). Thus, unlike the PAC tier—where clearance kinetics varied sharply by species (log-rank P ≈ 1 × 10⁻⁹)—aggregation at the PLC tier attenuates but does not entirely eliminate those differences, as reflected by the more modest global log-rank P value of 0.0397, which only just meets the conventional 0.05 threshold.
Patient-level clearance (PtLC)
Complete PtLC remained uncommon: 18 events occurred among 17 of 98 residents (18.4%), who achieved three consecutive negative cultures at all previously positive sites (Table 1). As shown in Fig. 1, one resident (patient 1004) reached PtLC twice—first after multi-MDRO clearance and again following re-isolation. The overall median time to PtLC was 97 d (95 % CI, 53–238; Fig. 2f), exceeding the pathogen-level medians for CRE (68 d), VRE (57 d), MRPA (26 d), and MRAB (28 d), as PtLC incorporates clearance across all four MDROs and thus cannot be shorter than any individual PLC.
Co-colonization profile
The upper panel of Table 3 shows patient counts. Among the 98 residents, CRE remained the dominant organism, colonizing 80 individuals (81.6%), whereas VRE, MRAB, and MRPA were present in 42.9%, 17.3%, and 19.4%, respectively. Co-carriage was common: 49 residents (50.0%) harbored at least two target MDROs, and 18 (18.4%) carried three or more, underscoring the polymicrobial pressure typical of long-term care. The most frequent dyad was CRE + VRE (27 of 98, 27.6%), followed by CRE + MRPA (16 of 98, 16.3%) and CRE + MRAB (13 of 98, 13.3%); no other pair exceeded 10% prevalence.
Notably, the lower panel of Table 3 presents a strongly asymmetric co-colonization network centered on CRE: 76.5 % of MRAB carriers and 84.2 % of MRPA carriers were also colonized with CRE, whereas the reverse probabilities were only 16.2 % and 20.0 %, respectively. For transparency, patient-level co-colonization profiles are provided in Supplementary Table S3.
Discussion
The present study refines our understanding of colonization dynamics in LTCFs by introducing a four-tier hierarchical framework—SAC, PAC, PLC, and PtLC—to disentangle clearance kinetics. At the PLC tier, organism‐specific differences in clearance are substantially attenuated but not eliminated (log-rank P = 0.0397), contrasting sharply with the highly significant differences observed at the PAC tier (log-rank P ≈ 1 × 10⁻⁹) and confirming that this additional stratification mitigates reservoir-driven confounding. This occurs because PLC has an intrinsic limitation: by aggregating sites and pathogens, it requires every previously culture-positive site to register three consecutive negative cultures. For example, CRE’s PLC can be substantially delayed by persistent stool colonization, even though sputum colonization clears much more quickly. To address this confounding, the PAC and SAC tiers were devised to focus separately on pathogen and site, respectively. The PAC tier captures intrinsic species behavior across sites, showing that some organisms (e.g., VRE) clear rapidly regardless of location, whereas others (e.g., CRE) persist. Prior reports confirm this pattern: 87.5% of VRE carriers had cleared compared with only 50% of CRE carriers [18], median ~26 weeks for VRE [19], while ~65% of CRE carriers remained colonized at one year [20]. These differences likely reflect ecological factors, as CRE persist within the colonic mucus layer whereas VRE are excluded [21]. The SAC tier provides a site-specific perspective: stool cultures clear extraordinarily slowly, reflecting the gut’s role as a protected reservoir where MDROs persist [6, 8], whereas sputum and urine often convert quickly because non-GI sites are more responsive to source control and local interventions [9, 10]—contrasts that PLC or PtLC would mask. For example, in patients with IDs 1023 and 1076, stool clearance was markedly delayed, while multiple urine sites cleared rapidly.
One might be tempted to attribute mortality to the colonizing pathogen, but when the intertwined confounding effects of anatomic site and species are disentangled, this assumption does not hold in an LTCF setting. PAD-tier survival curves for CRE, VRE, MRAB, and MRPA were nearly superimposable (log-rank P = 0.80; Fig. 2d), suggesting that no organism carried a significantly different mortality risk. This finding is plausible because LTCF residents are often chronically colonized with MDROs or merely isolated and quarantined without overt infection symptoms, meaning these pathogens contribute little to mortality. These results underscore that, in LTCFs, although organism-centered measures remain critical for transmission prevention and mortality prediction, improving survival hinges on addressing host factors (advanced age and male sex) and site-specific vulnerabilities—particularly sputum carriage—rather than solely targeting individual pathogens.
Patient attributes—age and sex—were significant predictors of mortality at both the SAD and PAD tiers. Although one might intuitively link age and sex to clearance, our study demonstrates the opposite: at both the SAC and PAC tiers, clearance was independent of these patient attributes, a distinction that can inform discussions with patients and caregivers—for example, those who wonder whether their elderly parent can be decolonized. This likely reflects that clearance is driven by local microbiological and environmental dynamics—biofilm architecture, microbiome composition, antibiotic pressure, and tissue comorbidities [8, 22, 23]—none of which correspond closely to systemic immunosenescence or sex-based immune differences.
This study has several limitations. First, our clearance definition was calibrated to Korean national regulations, which may limit generalizability to other settings. Second, follow-up began with the first positive culture at our facility, which may not represent the true onset of colonization; some residents were already colonized before admission, so carriage duration may have been underestimated. Third, universal baseline screening from negative through acquisition to clearance was not performed and was impractical in our LTCF setting: in Korea, isolation care is reimbursed fee-for-service, while non-isolation long-term care is largely reimbursed under a bundled payment system. Once acquisition is documented, caregivers expect serial cultures to enable isolation release, but they generally resist pre-acquisition screening because it could trigger quarantine, and hospitals must absorb the cost under bundled payment models. There is limited incentive on either side, making universal surveillance infeasible.
In this real-world context, automatic CRE screening after MRAB, MRPA, or VRE serves as a pragmatic alternative to universal surveillance. As shown in Table 3, co-colonization was strongly asymmetric. Because our conditional probabilities are longitudinal rather than contemporaneous at the trigger event, they do not themselves yield a cross-sectional number-needed-to-test (NNT). Nonetheless, more than three-quarters of MRAB carriers (76.5%) and over four-fifths of MRPA carriers (84.2%) also harbored CRE, whereas only 16.2% of CRE carriers had MRAB and 20.0% had MRPA. For facilities without active surveillance, this translates into targeted CRE screening with a single batched culture run for the flagged subset, reducing both nursing effort and laboratory costs compared with universal screening. Beyond this practical application, it remains essential to investigate directional dynamics more broadly: horizontal transfer of vanA from VRE to MRSA has produced de novo VRSA in co-colonized patients [24]. Such cases demonstrate how simultaneous colonization within the same niche facilitates gene transfer, underscoring the directional and asymmetric nature of these interactions.
In conclusion, this study offers a distinctive contribution by pairing a four-tier hierarchical framework with conditional-probability mapping to disentangle the often-confounded influences of pathogen and anatomic site in LTCFs—an angle rarely explored in previous MDRO research. It overturns intuitive expectations by showing that, in this LTCF, mortality depends more on colonization site than on pathogen identity (MDROs here are often detected as chronic colonizers rather than causing acute illness), whereas clearance is independent of patient attributes such as age and sex—a finding that may reassure caregivers worried that advanced age impedes decolonization. The asymmetric co-colonization pattern we uncovered—non-CRE organisms almost always co-colonized with CRE, whereas CRE carriers rarely host other MDROs—supports automatic CRE screening whenever MRPA, MRAB, or VRE is detected and further highlights the need for systematic investigation of directional co-colonization dynamics—such as transfer of vanA from VRE to MRSA leading to de novo VRSA, thereby increasing P(VRSA | VRE + MRSA)—in LTCFs burdened by multiple MDROs.
References
[1] EclinicalMedicine: Antimicrobial resistance: a top ten global public health threat. EClinicalMedicine 2021;41: 101221.
[2] O'Fallon E, Pop-Vicas A, D'Agata E: The emerging threat of multidrug-resistant gram-negative organisms in long-term care facilities. J Gerontol A Biol Sci Med Sci 2009;64(1): 138-141.
[3] Centers for Disease C, Prevention: Vital signs: carbapenem-resistant Enterobacteriaceae. MMWR Morbidity and mortality weekly report 2013;62(9): 165-170.
[4] Cilloniz C, Calabretta D, Palomeque A, Gabarrus A, Ferrer M, Marcos MA, et al.: Risk Factors and Outcomes Associated With Polymicrobial Infection in Community-Acquired Pneumonia. Arch Bronconeumol 2025.
[5] Tadese BK, DeSantis SM, Mgbere O, Fujimoto K, Darkoh C: Clinical Outcomes Associated with Co-infection of Carbapenem-Resistant Enterobacterales and other Multidrug-Resistant Organisms. Infect Prev Pract 2022;4(4): 100255.
[6] Mo Y, Hernandez-Koutoucheva A, Musicha P, Bertrand D, Lye D, Ng OT, et al.: Duration of Carbapenemase-Producing Enterobacteriaceae Carriage in Hospital Patients. Emerg Infect Dis 2020;26(9): 2182-2185.
[7] Nutman A, Lerner A, Fallach N, Schwartz D, Carmeli Y: Likelihood of persistent carriage of carbapenem-resistant Acinetobacter baumannii on readmission in previously identified carriers. Infect Control Hosp Epidemiol 2019;40(10): 1188-1190.
[8] Byers KE, Anglim AM, Anneski CJ, Farr BM: Duration of colonization with vancomycin-resistant Enterococcus. Infect Control Hosp Epidemiol 2002;23(4): 207-211.
[9] Haverkate MR, Derde LP, Brun-Buisson C, Bonten MJ, Bootsma MC: Duration of colonization with antimicrobial-resistant bacteria after ICU discharge. Intensive Care Med 2014;40(4): 564-571.
[10] Pacio GA, Visintainer P, Maguire G, Wormser GP, Raffalli J, Montecalvo MA: Natural history of colonization with vancomycin-resistant enterococci, methicillin-resistant Staphylococcus aureus, and resistant gram-negative bacilli among long-term-care facility residents. Infect Control Hosp Epidemiol 2003;24(4): 246-250.
[11] Lin I-W, Huang C-Y, Pan S-C, Chen Y-C, Li C-M: Duration of colonization with and risk factors for prolonged carriage of multidrug resistant organisms among residents in long-term care facilities. Antimicrobial Resistance & Infection Control 2017;6.
[12] Zirakzadeh A, Patel R: Vancomycin-resistant enterococci: colonization, infection, detection, and treatment. Mayo Clin Proc 2006;81(4): 529-536.
[13] Heinze K, Kabeto M, Martin ET, Cassone M, Hicks L, Mody L: Predictors of methicillin-resistant Staphylococcus aureus and vancomycin-resistant enterococci co-colonization among nursing facility patients. Am J Infect Control 2019;47(4): 415-420.
[14] Institute CaLS. Performance Standards for Antimicrobial Susceptibility Testing. Wayne, PA. 2023.
[15] Lortholary O, Fagon JY, Buu Hoi A, Mahieu G, Gutmann L: Colonization by Acinetobacter baumanii in intensive-care-unit patients. Infect Control Hosp Epidemiol 1998;19(3): 188-190.
[16] Estepa V, Rojo-Bezares B, Torres C, Saenz Y: Faecal carriage of Pseudomonas aeruginosa in healthy humans: antimicrobial susceptibility and global genetic lineages. FEMS Microbiol Ecol 2014;89(1): 15-19.
[17] Kleinbaum DG, Klein M. Survival Analysis: A self-Learning Text. LLC, 233 Spring Street, New York, NY 10013, USA: Springer Science + Business Media. 2012.
[18] Dinh A, Fessi H, Duran C, Batista R, Michelon H, Bouchand F, et al.: Clearance of carbapenem-resistant Enterobacteriaceae vs vancomycin-resistant enterococci carriage after faecal microbiota transplant: a prospective comparative study. J Hosp Infect 2018;99(4): 481-486.
[19] Shenoy ES, Paras ML, Noubary F, Walensky RP, Hooper DC: Natural history of colonization with methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococcus (VRE): a systematic review. BMC Infect Dis 2014;14: 177.
[20] Bar-Yoseph H, Hussein K, Braun E, Paul M: Natural history and decolonization strategies for ESBL/carbapenem-resistant Enterobacteriaceae carriage: systematic review and meta-analysis. J Antimicrob Chemother 2016;71(10): 2729-2739.
[21] Caballero S, Carter R, Ke X, Susac B, Leiner IM, Kim GJ, et al.: Distinct but Spatially Overlapping Intestinal Niches for Vancomycin-Resistant Enterococcus faecium and Carbapenem-Resistant Klebsiella pneumoniae. PLoS Pathog 2015;11(9): e1005132.
[22] Ciobotaro P, Flaks-Manov N, Oved M, Schattner A, Hoshen M, Ben-Yosef E, et al.: Predictors of Persistent Carbapenem-Resistant Enterobacteriaceae Carriage upon Readmission and Score Development. Infect Control Hosp Epidemiol 2016;37(2): 188-196.
[23] Seong H, Lee SK, Cheon JH, Yong DE, Koh H, Kang YK, et al.: Fecal Microbiota Transplantation for multidrug-resistant organism: Efficacy and Response prediction. J Infect 2020;81(5): 719-725.
[24] Marchaim D, Perez F, Lee J, Bheemreddy S, Hujer AM, Rudin S, et al.: "Swimming in resistance": Co-colonization with carbapenem-resistant Enterobacteriaceae and Acinetobacter baumannii or Pseudomonas aeruginosa. Am J Infect Control 2012;40(9): 830-835.
Pulmonary resection improves survival when combined with optimized MDR-TB chemotherapy
Surgical candidates benefit most when sputum conversion is incomplete or disease burden is extensive
Different resection types (wedge, segmentectomy, lobectomy) demonstrate comparable mortality when appropriately selected
Adverse events remain acceptable when surgery is performed in high-volume thoracic centers
Meta-analytic evidence supports surgery as an adjunct—not replacement—to standard MDR-TB medical therapy
Piperacillin-tazobactam versus meropenem for ceftriaxone-resistant Enterobacterales bacteremia: visual synthesis across key studies
The clearest head-to-head comparison for piperacillin-tazobactam (pip/tazo) in ceftriaxone-resistant E. coli or K. pneumoniae bacteremia comes from a randomized trial (MERINO), in which 30-day mortality was higher with pip/tazo than with meropenem.
Earlier observational cohorts comparing carbapenems with pip/tazo or broader BLBLI therapy showed mixed signals, largely in parallel with substantial differences in baseline severity and infection source distribution.
I. At-a-glance dashboard
Central head-to-head question
Pip/tazo vs meropenem
Ceftriaxone-resistant E. coli or K. pneumoniae bacteremia (randomized trial)
Primary mortality signal (MERINO)
+8.6% absolute
30-day mortality: 12.3% vs 3.7% (pip/tazo minus meropenem)
Observational context (two cohorts)
Mixed
Adjusted hazard ratio estimates ranged from neutral to harmful for BLBLI/pip-tazo vs carbapenems
Study (year)
Design focus
Comparator framing
Main pip/tazo (or BLBLI) signal
Best single takeaway
Harris (MERINO), 2018
Randomized clinical trial
Pip/tazo vs meropenem
Higher mortality with pip/tazo
Meropenem demonstrated a materially lower 30-day mortality than pip/tazo in the randomized comparison.
Tamma, 2015
Retrospective cohort (higher acuity)
Pip/tazo vs carbapenem
Harm signal for pip/tazo
Adjusted results suggested higher hazard of death with pip/tazo in a higher-severity cohort.
Rodriguez-Baño, 2012
Retrospective cohort (lower acuity)
BLBLI vs carbapenem
No detected harm (wide CI)
In a predominantly low-severity, urinary/biliary-source cohort, BLBLI definitive therapy did not show an excess mortality signal after adjustment.
II. Central evidence: Harris (MERINO) randomized trial emphasizing pip/tazo versus meropenem
Primary endpoint (30-day mortality)
Primary analysis: pip/tazo 23/187 (12.3%) vs meropenem 7/191 (3.7%).
Absolute risk difference: +8.6% (pip/tazo minus meropenem), approximately 1 additional death per ~12 treated (derived from the displayed counts).
30-day mortality (primary analysis) — bar lengths scaled to 15%
Piperacillin-tazobactam
12.3% (23/187)
Meropenem
3.7% (7/191)
Visual scaling note: 15% corresponds to 100% bar width to improve legibility of low event rates.
30-day mortality (per-protocol) — bar lengths scaled to 15%
Piperacillin-tazobactam
10.6% (18/170)
Meropenem
3.8% (7/186)
These two panels show consistent directionality (higher mortality with pip/tazo) across analytic sets.
Secondary endpoints displayed in the trial summary table
The displayed endpoints include early clinical/microbiological success and specific failure events (microbiological relapse; secondary infection with multidrug-resistant organisms or C. difficile).
Directional differences favored meropenem for early success metrics and favored meropenem for failure-event rates, with confidence intervals presented as broad for several endpoints.
Endpoint (as displayed)
Pip/tazo
Meropenem
Between-group difference (pip/tazo − meropenem)
Clinical and microbiological success at day 4
121/177 (68.4%)
138/185 (74.6%)
-6.2% (95% CI -15.5 to 3.1)
Microbiological success at day 4
169/174 (97.1%)
184/185 (99.5%)
-2.3% (95% CI -6.1 to 0.4)
Microbiological relapse
9/187 (4.8%)
4/191 (2.1%)
+2.7% (95% CI -1.1 to 7.1)
Secondary infection with multidrug-resistant organism or C. difficile
15/187 (8.0%)
8/191 (4.2%)
+3.8% (95% CI -1.1 to 9.1)
III. Observational comparison to support interpretation: Tamma (2015) and Rodriguez-Baño (2012)
Effect-size visualization (adjusted hazard ratios versus carbapenems)
The following graphic summarizes adjusted hazard ratio point estimates and confidence intervals shown in the slide tables, using a shared scale from 0 to 4.
The vertical reference line marks HR = 1.0 (no difference); points to the right suggest higher hazard with pip/tazo or BLBLI therapy, while points to the left suggest lower hazard.
01234
Rodriguez-Baño 2012 (BLBLI vs carbapenem)
HR 0.76 (0.28–2.07)
Tamma 2015 (pip/tazo vs carbapenem)
HR 1.92 (1.07–3.45)
Visual scaling note: the axis is fixed at 0–4 for both rows; confidence intervals extending beyond the axis would be truncated (not applicable to the displayed values).
Case-mix contrasts that plausibly drive discordant observational signals
Rodriguez-Baño cohort: predominantly E. coli, urinary/biliary sources ~68%, Pitt score median ~1, ICU admission ~8.7%, neutropenia ~4.8%.
Tamma cohort: mixed pathogens (E. coli, K. pneumoniae, Proteus), urinary/biliary sources ~30%, Pitt score mean ~2.2, ICU admission ~33.8%, neutropenia ~15%.
These contrasts align with a common pattern in severe infection therapeutics: higher acuity and less favorable sources increase the clinical penalty of marginal pharmacodynamic performance.
Dimension
Rodriguez-Baño 2012 (lower acuity profile)
Tamma 2015 (higher acuity profile)
Interpretive implication
Organisms
Primarily E. coli
E. coli, K. pneumoniae, Proteus
Greater heterogeneity may shift MIC distributions and reduce the efficacy margin for pip/tazo.
Urinary/biliary source proportion
~68%
~30%
More non-urinary sources may correlate with higher inoculum and more complex source control.
Baseline severity
Pitt median ~1; ICU ~8.7%
Pitt mean ~2.2; ICU ~33.8%
Effect modification by acuity can amplify an empirical advantage for carbapenems.
Neutropenia
~4.8%
~15%
Immunosuppression increases consequence of delayed or insufficient bactericidal exposure.
Adjusted treatment estimate
BLBLI: HR 0.76 (0.28–2.07)
Pip/tazo: HR 1.92 (1.07–3.45)
Opposing point estimates appear consistent with different case-mix rather than a true contradiction.
IV. Integrated conclusion centered on pip/tazo head-to-head evidence
What the evidence most directly suggests about pip/tazo versus meropenem
The randomized MERINO comparison showed higher 30-day mortality with pip/tazo than with meropenem in ceftriaxone-resistant E. coli or K. pneumoniae bacteremia.
Within the displayed secondary outcomes, early success favored meropenem directionally, while relapse and secondary infection rates were numerically higher with pip/tazo (with wide intervals for several endpoints).
How the observational landscape fits around the randomized signal
Observational cohorts showed mixed results: a low-acuity, source-favorable cohort appeared compatible with no excess mortality using BLBLI definitive therapy, whereas a higher-acuity cohort showed an adjusted harm signal for pip/tazo.
The randomized trial aligns more closely with the higher-acuity harm-signal direction than with the low-acuity neutral-signal direction.
For ceftriaxone-resistant or ESBL-grade bacteremia, meropenem (carbapenem therapy) appears to have the most robust supportive signal when a direct comparison to pip/tazo is prioritized.
Any carbapenem-sparing use of pip/tazo or other BLBLI therapy appears most defensible only in carefully selected, lower-risk patterns resembling the low-acuity observational cohort case-mix (favorable sources and clinical stability), while acknowledging that randomized evidence has demonstrated meaningful mortality separation in the head-to-head setting.
This synthesis is intended for educational discussion; individual management decisions should reflect patient-specific severity, infection source, microbiologic details, dosing strategy, and local policies.
Abbreviations
pip/tazo: piperacillin-tazobactam
BLBLI: β-lactam/β-lactamase inhibitor
ESBL: extended-spectrum β-lactamase
CI: confidence interval
HR: hazard ratio
V. References
Harris PNA, Tambyah PA, Lye DC, et al. Effect of piperacillin-tazobactam vs meropenem on 30-day mortality for patients with E. coli or K. pneumoniae bloodstream infection and ceftriaxone resistance: a randomized clinical trial. JAMA. 2018;320:984-994.
Tamma PD, Han JH, Rock C, et al. Carbapenem therapy is associated with improved survival compared with piperacillin-tazobactam for patients with extended-spectrum β-lactamase bacteremia. Clinical Infectious Diseases. 2015;60:1319-1325.
Rodríguez-Baño J, Navarro MD, Retamar P, et al. β-Lactam/β-lactamase inhibitor combinations for the treatment of bloodstream infections due to extended-spectrum β-lactamase–producing Escherichia coli. Clinical Infectious Diseases. 2012;54:167-174.
Survival Function \( S(t) \) is defined as:
\[
S(t) = P(T > t),
\]
where \( T \) is the random variable representing the time to event (clearance). \( S(t) \) describes the probability that a subject remains non-cleared (has not yet achieved three consecutive negative tests) after time \( t \).
Hazard Function \( \lambda(t) \) is defined as:
\[
\lambda(t) = \lim_{\Delta t \to 0} \frac{P(t \le T < t + \Delta t \mid T \ge t)}{\Delta t}.
\]
It represents the instantaneous rate of achieving clearance at time \( t \), given that the patient has not cleared the pathogen before \( t \).
1.2. Kaplan-Meier Estimator
The Kaplan-Meier (KM) estimator is a non-parametric statistic used to estimate the survival function \( S(t) \). If there are \( n \) subjects, let \( t_{(1)}, t_{(2)}, \dots, t_{(k)} \) be the distinct event times in ascending order. Let \( d_j \) be the number of events (clearances) at time \( t_{(j)} \) and \( r_j \) be the number of subjects at risk just before \( t_{(j)} \). The KM estimator is:
\( \lambda_0(t) \) is the baseline hazard function (unspecified),
\( \mathbf{x}_i \) is the vector of covariates (e.g., site, age, gender),
\( \boldsymbol{\beta} \) is the vector of coefficients.
A hazard ratio between two covariate levels reflects the ratio of their hazards at any given time \( t \). If a coefficient \( \beta_j \) is positive, it suggests an increased rate of clearance for that factor level.
1.4. Fine-Gray Model for Competing Risks
When multiple competing events can occur (e.g., clearance is the event of interest, but death or loss to follow-up might preclude clearance), a competing risk model such as Fine and Gray is used:
\[
\text{Fine-Gray: } \quad \text{Subdistribution hazard } = h_j(t) = \lim_{\Delta t \to 0}
\frac{P(t \le T < t + \Delta t, \epsilon = j \mid T \ge t \text{ or } (T < t \text{ and } \epsilon \neq j))}{\Delta t},
\]
where \( \epsilon \) indicates the type of event (\( j = \) clearance, other = competing events). This method accounts for the fact that once a competing event (e.g., death) happens, one can no longer clear the pathogen.
2. Code and Step-by-Step Explanation
Step
Section
Purpose
0
Install Packages
Ensures necessary packages are installed
1
Define Directory
Output directory for storing analysis results
2
Data Import/Prep
Loads and cleans the data
3
Exploratory Analysis
Summaries and missing data checks
4
Kaplan-Meier Analysis
Non-parametric survival estimates by site
5
Cox Model (Simple)
Estimates hazard ratios for clearance with Site only
6
Multicollinearity Check
Detects correlated predictors using VIF
7
Penalized Cox (Lasso)
Regularization to handle many or correlated predictors
8
Fine-Gray Model
Competing risks approach for clearance vs. death
9
Visualization
Visual compares survival curves by site
10
Recommendations/Checks
Influential points, outliers, and missing data
11
End of Script
Wrap-up, optional housekeeping
Note: The code presented here is R-based and uses packages such as survival, survminer, cmprsk, dplyr, ggplot2, and glmnet. Adjust package calls as needed for local environment.
Interpretation:
- This section ensures that all required packages for reading data, plotting, modeling, and statistical testing are present and loaded.
- Automated checks help avoid missing dependencies during runtime.
Interpretation:
- Specifies where plots and model summaries will be stored.
- Recursive creation of directories ensures sub-directories exist when writing outputs.
2.2. Data Import and Preparation
# 2. Data Import and Preparation
data_path <- file.path(desktop_path, "dataset05.csv")
my_data <- read_csv(data_path, locale = locale(encoding = "UTF-8"))
cat("First few rows of the dataset:\n")
print(head(my_data))
cat("Structure of the dataset:\n")
print(str(my_data))
# Convert variables to appropriate data types
my_data <- my_data %>%
mutate(
PatientID = as.factor(PatientID),
Pathogen = as.factor(Pathogen),
Site = as.factor(Site),
Start_Date = as.Date(Start_Date, format = "%m/%d/%Y"),
Event_Date = as.Date(Event_Date, format = "%m/%d/%Y"),
Event_Type = as.factor(Event_Type),
Event_Code = as.numeric(Event_Code),
Age = as.numeric(Age),
Gender = as.factor(Gender),
FactorA = as.factor(FactorA),
FactorB = as.factor(FactorB),
FactorC = factor(FactorC, levels = c("Low", "Medium", "High"), ordered = TRUE)
)
cat("Structure after type conversions:\n")
print(str(my_data))
Interpretation:
- Reads the CSV file containing survival (clearance) data and sets correct data types for key variables.
- Event_Code should indicate whether an event (clearance) occurred (e.g., 1 = clearance, 0 = censored, 2 = competing event if used).
- This step ensures subsequent survival analysis is consistent with R’s expectations for times, events, and factors.
2.3. Exploratory Data Analysis (EDA)
# 3. Exploratory Data Analysis (EDA)
cat("Summary statistics:\n")
print(summary(my_data))
cat("Missing values per column:\n")
print(sapply(my_data, function(x) sum(is.na(x))))
cat("Event distribution across sites:\n")
print(table(my_data$Site, my_data$Event_Code))
Interpretation:
- Summary statistics and missing value checks identify potential data issues.
- A two-way table with Site vs. Event_Code reveals how many clearance events occurred at each site.
2.4. Kaplan-Meier Plots Stratified by Site
# 4. Kaplan-Meier Plots Stratified by Site
if (!"Time_to_Event" %in% colnames(my_data)) {
my_data <- my_data %>%
mutate(Time_to_Event = as.numeric(difftime(Event_Date, Start_Date, units = "days")))
}
if (any(my_data$Time_to_Event <= 0, na.rm = TRUE)) {
cat("There are observations with non-positive Time_to_Event. These will be removed.\n")
my_data <- my_data %>%
filter(Time_to_Event > 0)
}
# Create the survival object
surv_object_site <- Surv(time = my_data$Time_to_Event, event = my_data$Event_Code == 1)
# Fit Kaplan-Meier survival curves stratified by Site
km_fit_site <- survfit(surv_object_site ~ Site, data = my_data)
# Plot the Kaplan-Meier curves
km_plot <- ggsurvplot(
km_fit_site,
data = my_data,
risk.table = TRUE,
pval = TRUE, # Adds p-value from log-rank test
conf.int = TRUE,
xlab = "Time in Days",
ylab = "Probability of Clearance",
title = "Kaplan-Meier Curves Stratified by Site",
legend.title = "Site",
palette = scales::hue_pal()(length(levels(my_data$Site)))
)
print(km_plot)
ggsave(
filename = file.path(output_dir, "Kaplan_Meier_Curves_Stratified_by_Site.png"),
plot = km_plot$plot, width = 8, height = 6
)
ggsave(
filename = file.path(output_dir, "Kaplan_Meier_Risk_Table_Stratified_by_Site.png"),
plot = km_plot$table, width = 8, height = 3
)
Mathematical Note:
- The Kaplan-Meier approach estimates \( \hat{S}(t) \) (the probability that clearance has not yet occurred by time \( t \)).
- Stratification by Site compares different clearance curves. A log-rank test checks if the curves are significantly different.
Interpretation:
- A steeper curve indicates faster clearance.
- The p-value clarifies whether differences in clearance times across sites are statistically significant.
2.5. Cox Proportional Hazards Model Including Site
# 5. Cox Proportional Hazards Model Including Site
cox_model_simple <- coxph(surv_object_site ~ Site, data = my_data)
cat("Summary of Simplified Cox Model (Only Site):\n")
summary_cox_simple <- summary(cox_model_simple)
print(summary_cox_simple)
sink(file = file.path(output_dir, "Summary_Simplified_Cox_Model.txt"))
print(summary_cox_simple)
sink()
# Test proportional hazards assumption
ph_test_simple <- tryCatch(
cox.zph(cox_model_simple),
error = function(e) {
cat("Error in proportional hazards test:\n", e$message, "\n")
return(NULL)
}
)
if (!is.null(ph_test_simple)) {
cat("Proportional Hazards Test for Simplified Model:\n")
print(ph_test_simple)
sink(file = file.path(output_dir, "PH_Test_Simplified_Cox_Model.txt"))
print(ph_test_simple)
sink()
ph_plot_simple <- ggcoxzph(ph_test_simple)
print(ph_plot_simple)
ggsave(
filename = file.path(output_dir, "Schoenfeld_Residuals_Simplified_Cox_Model.png"),
plot = ph_plot_simple, width = 8, height = 6
)
} else {
cat("Proportional hazards assumption test could not be performed.\n")
}
Mathematical Note:
- The Cox model: \( \lambda_i(t) = \lambda_0(t) \exp(\beta_1 X_{i1} + \cdots + \beta_p X_{ip}) \).
- Here, the predictor is only Site (categorical). The exponent of a coefficient, \( \exp(\hat{\beta}) \), is the hazard ratio.
- If \( \exp(\hat{\beta}) > 1 \), that site experiences a faster clearance (on average).
- If \( \exp(\hat{\beta}) < 1 \), that site has a slower clearance rate compared to the reference site.
Interpretation:
- Cox.ZPH test checks whether proportional hazards assumption is valid.
- If non-significant, it suggests no major violation of the assumption for that predictor.
2.6. Checking for Multicollinearity
# 6. Checking for Multicollinearity
cox_model_full <- coxph(surv_object_site ~ Site + FactorA + FactorB + FactorC + Age + Gender, data = my_data)
print("Summary of Full Cox Model:")
summary_cox_full <- summary(cox_model_full)
print(summary_cox_full)
sink(file = file.path(output_dir, "Summary_Full_Cox_Model.txt"))
print(summary_cox_full)
sink()
# Compute VIF
if(!is.null(cox_model_full$coefficients)) {
design_matrix <- model.matrix(~ Site + FactorA + FactorB + FactorC + Age + Gender, data = my_data)[, -1]
lm_fit <- lm(Time_to_Event ~ ., data = as.data.frame(design_matrix))
vif_values <- vif(lm_fit)
print("Variance Inflation Factor (VIF) for Predictors:")
print(vif_values)
write.csv(as.data.frame(vif_values), file = file.path(output_dir, "VIF_Full_Cox_Model.csv"), row.names = TRUE)
} else {
warning("Full Cox model did not converge. Skipping VIF computation.")
}
Mathematical Note:
- VIF (Variance Inflation Factor) for each predictor \( X_j \) is:
\[
\mathrm{VIF}_j = \frac{1}{1 - R_j^2},
\]
where \( R_j^2 \) is the coefficient of determination when \( X_j \) is regressed on all other predictors.
- High VIF (commonly > 5 or 10) indicates multicollinearity.
Interpretation:
- Identifying collinear factors ensures stable model estimates.
- High collinearity in a Cox model can lead to large standard errors for coefficient estimates.
Mathematical Note:
- Penalized Cox adds an \( L_1 \) penalty term \( \alpha \lambda \|\beta\|_1 \) (when \( \alpha=1 \)) to the partial likelihood objective. This is known as Lasso, encouraging sparse solutions (some coefficients shrink to 0).
Interpretation:
- Lasso helps in variable selection and reducing overfitting, especially when many predictors might be collinear or the sample size is small.
2.8. Fine-Gray Model for Competing Risks
# 8. Fine-Gray Model for Competing Risks
# Event_Code == 1: Clearance (event of interest)
# Event_Code == 2: Some competing event (e.g., Death)
my_data_fg <- my_data %>%
mutate(
Competing_Event = case_when(
Event_Code == 1 ~ 0, # Event of interest
Event_Code == 2 ~ 1, # Competing event
TRUE ~ NA_real_
)
)
na_count_fg <- sum(is.na(my_data_fg$Competing_Event))
print(paste("Number of NA values in Competing_Event:", na_count_fg))
write.csv(data.frame(Column = "Competing_Event", NA_Count = na_count_fg),
file = file.path(output_dir, "NA_Counts_Competing_Event.csv"),
row.names = FALSE)
my_data_clean_fg <- my_data_fg %>%
filter(!is.na(Competing_Event)) %>%
drop_na(Site, FactorA, FactorB, FactorC, Age, Gender, Time_to_Event, Competing_Event)
print("Dimensions after cleaning for Fine-Gray model:")
print(dim(my_data_clean_fg))
write.csv(data.frame(Dimensions = paste(dim(my_data_clean_fg), collapse = "x")),
file.path(output_dir, "Fine_Gray_Clean_Data_Dimensions.csv"),
row.names = FALSE)
covariates_fg <- model.matrix(~ Site + FactorA + FactorB + FactorC + Age + Gender, data = my_data_clean_fg)[, -1]
print(paste("Number of covariates:", ncol(covariates_fg)))
print(paste("Number of observations:", nrow(my_data_clean_fg)))
write.csv(data.frame(Covariates = ncol(covariates_fg), Observations = nrow(my_data_clean_fg)),
file.path(output_dir, "Covariate_Matrix_Dimensions_Fine_Gray.csv"),
row.names = FALSE)
if(nrow(covariates_fg) == nrow(my_data_clean_fg)) {
fg_model_site <- try(crr(
ftime = my_data_clean_fg$Time_to_Event,
fstatus = my_data_clean_fg$Competing_Event,
cov1 = covariates_fg
), silent = TRUE)
if(class(fg_model_site) != "try-error") {
print("Summary of Fine-Gray Model:")
summary_fg <- summary(fg_model_site)
print(summary_fg)
sink(file = file.path(output_dir, "Summary_Fine_Gray_Model.txt"))
print(summary_fg)
sink()
} else {
warning("Fine-Gray model failed to converge. Check data for issues.")
error_message <- "Fine-Gray model failed to converge. Check data for issues."
write.csv(data.frame(Error = error_message),
file.path(output_dir, "Fine_Gray_Model_Error.csv"),
row.names = FALSE)
}
} else {
stop("Mismatch in the number of rows between covariates and survival data for Fine-Gray model.")
}
Mathematical Note:
- Fine and Gray (1999) proposed modeling the subdistribution hazard of a particular event type in the presence of competing events.
- The subdistribution hazard function is different from the cause-specific hazard: it explicitly accounts for individuals who have experienced the competing event but remain “at risk” in the subdistribution sense.
Interpretation:
- Use this approach if competing events (like death) might preclude observing clearance.
- The model yields subdistribution hazard ratios, interpreted as the effect of covariates on the cumulative incidence of clearance.
2.9. Visualizing Survival Differences Across Sites
Interpretation:
- Faceted or combined survival plots highlight clearance patterns for each site.
- This step is crucial for visual diagnosis of clearance timelines across multiple categories.
2.10. Additional Recommendations and Checks
# 10. Additional Recommendations and Checks
# 10.1. Check for Influential Observations
influence_cox <- residuals(cox_model_simple, type = "dfbeta")
if(!is.null(influence_cox)) {
dfbeta_df <- as.data.frame(influence_cox)
dfbeta_df$PatientID <- rownames(dfbeta_df)
library(reshape2)
dfbeta_melt <- melt(dfbeta_df, id.vars = "PatientID")
dfbeta_plot <- ggplot(dfbeta_melt, aes(x = PatientID, y = value, color = variable)) +
geom_point() +
geom_hline(yintercept = 0, linetype = "dashed") +
labs(title = "DFBeta for Simplified Cox Model", x = "Patient ID", y = "DFBeta") +
theme_minimal() +
theme(axis.text.x = element_text(angle = 90, vjust = 0.5))
print(dfbeta_plot)
ggsave(filename = file.path(output_dir, "DFBeta_Simplified_Cox_Model.png"),
plot = dfbeta_plot, width = 12, height = 6)
} else {
warning("No dfbeta available for the simplified Cox model.")
}
# 10.2. Assess Outliers in Continuous Variables
age_boxplot <- ggplot(my_data, aes(x = "", y = Age)) +
geom_boxplot(fill = "#2E9FDF") +
labs(title = "Boxplot of Age", y = "Age") +
theme_minimal()
print(age_boxplot)
ggsave(filename = file.path(output_dir, "Boxplot_Age.png"),
plot = age_boxplot, width = 6, height = 6)
# 10.3. Handling Missing Data
# Consider advanced imputation if missingness is not negligible.
Interpretation:
- DFBeta plots: Identifies influential points that might unduly affect estimates.
- Boxplot of Age: Quickly spot outliers or unusual distributions.
2.11. End of Script
# 11. End of Script
# Most plots and model summaries have already been saved in previous steps.
# Additional saves can be added if needed.
Written on December 24th, 2024
MDRO R Scripts
SAC_SAD.R
PAC_PAD.R
PLC_PtLC.R
Co-occurance.R
Federated Learning Server (Under Development)
Federated Learning: Symbiotic Algorithm for Privacy-Preserving Collaboration
In the context of Web 4.0, Federated Learning becomes an indispensable tool in the exploration and development of privacy-preserving machine learning models. This approach aligns seamlessly with the principles of symbiotic interaction within Web 4.0, facilitating the creation of machine learning interfaces that leverage decentralized data without compromising individual privacy. By incorporating Federated Learning, the project enhances its ability to process sensitive data across multiple decentralized sources—such as user interactions across various web platforms—while ensuring that personal data remains on the user's device. This method not only respects user privacy but also enriches the machine learning models with a diverse range of data inputs, leading to more personalized and efficient user experiences.
Moreover, the integration of Federated Learning within Web 4.0 frameworks underscores a significant shift towards more ethical and user-centric design philosophies. It exemplifies how advanced technologies can be harnessed to foster collaborative advancement, protect individual privacy, and enhance the collective intelligence of the system without centralizing personal data. This approach contributes to the development of a more secure, efficient, and privacy-preserving Web 4.0 ecosystem, where machine learning models are continually refined and improved through encrypted, aggregated updates shared across devices. Thus, Federated Learning stands as a cornerstone in the advancement of exploratory machine learning for symbiotic Web 4.0, embodying the project's commitment to privacy, collaboration, and innovation.
Federated Learning Platform for Multi-Drug Resistance Research (Under Dev)
The need for a "Federated Learning Platform for Multi-Drug Resistance Research" arises from the pressing challenges posed by multi-drug resistant infections and the critical importance of maintaining patient privacy while harnessing data across multiple healthcare institutions. Federated learning, as described, enables multiple hospitals to contribute to a joint research initiative without the need to share raw patient data. This decentralized approach aligns perfectly with the sensitive nature of handling medical records and the logistical complexities of multi-institutional research.
Benefits of Federated Learning in Multi-Drug Resistance Research:
Privacy Preservation: In line with GDPR guidelines, federated learning ensures that each participating hospital trains models on its own data locally. This means that sensitive patient information does not leave the hospital premises, significantly reducing the risk of data breaches and unauthorized access.
Enhanced Collaborative Research: By allowing each institution to contribute only model updates rather than raw data, federated learning facilitates a collaborative environment without compromising the integrity of individual data sets. This is particularly crucial in studies involving multi-drug resistance, where data from diverse demographics and treatment outcomes is vital for robust research findings.
Scalability and Flexibility: The federated learning platform can easily scale to include more participants over time, enhancing the model's accuracy and predictive power as more data points are indirectly shared. This scalability ensures that the research remains adaptable to new data without needing constant system overhauls.
Local Processing with Global Improvements: Each hospital can benefit from collective improvements in the model while ensuring that the local data used to train these models reflects their specific patient population's characteristics. This local-global approach is especially advantageous in studying varied responses to drug treatments across different populations.
Further Research
Markov and Hidden Markov Chain Analysis of Pathogen Clearance Dynamics (Written October 25, 2025)
I. Introduction
Pathogen colonization in environments such as healthcare facilities can persist for extended periods before the host successfully clears the organism. For example, residents in long-term care facilities often carry antibiotic-resistant bacteria on their skin or in their mucosa for weeks or even months. Understanding the clearance dynamics of such colonization is crucial: it informs infection control policies (such as isolation duration and decolonization strategies) and helps predict the risk of ongoing transmission. However, quantifying clearance is challenging because detection of pathogens may be imperfect (due to intermittent sampling or test limitations) and individuals may become re-colonized after clearance.
Markov chain models provide a powerful framework for analyzing longitudinal infection status data. By treating colonization and clearance as probabilistic state transitions, a Markov model can estimate the chance an individual will clear an infection in a given time interval based on observational data. This approach leverages the memoryless property: the future risk of clearance or persistence depends only on the current state of colonization, not the history. It thereby simplifies complex temporal processes into tractable probabilities. Moreover, Markov models can accommodate recurrent events (re-colonization) naturally by allowing transitions back and forth between states.
A limitation of basic Markov models in this context is the assumption that the infection state is directly observed without error. In practice, diagnostic tests for colonization (e.g., cultures or PCR) are not 100% sensitive, meaning a colonized individual might occasionally test negative while still harboring the pathogen. To address this, we can extend the model to a Hidden Markov Model (HMM), wherein the underlying infection status is a hidden (latent) state that evolves via a Markov chain, and the observed test results are probabilistic signals of that true state. The HMM framework allows estimation of both the transition probabilities (colonization and clearance rates) and the accuracy of the observations (sensitivity and specificity of tests).
In this study, we develop and apply both a Markov chain model and a Hidden Markov model to a longitudinal dataset of pathogen colonization among facility residents. The dataset, published previously as a supplementary file in an earlier study, includes weekly screening results for various pathogens (e.g., multidrug-resistant bacteria) and body sites. Due to limited sample size for any single pathogen–site combination, all observations were pooled to estimate an overall clearance model – effectively assuming that, regardless of pathogen species or colonization site, the clearance dynamics follow a similar pattern. Within this framework, we estimate key parameters such as the weekly probability of clearance and acquisition (new colonization), and evaluate the capability of the model to predict clearance in future cases. The inclusion of the HMM approach enables a more robust analysis by accounting for possible misclassification of colonization status. The ultimate goal is to establish a probabilistic model of clearance that can be used to forecast outcomes and guide decision-making in infection control.
II. Method
The analysis was conducted in discrete time with one-week intervals, corresponding to the weekly screening schedule of the study. All residents in the dataset were assumed to follow the same transition probabilities for colonization and clearance, meaning individual differences and pathogen-specific effects were not explicitly modeled. This homogeneous assumption was necessitated by data limitations and provides a first-order approximation of the clearance process.
A. Data and Assumptions
The dataset consisted of weekly observations of colonization status for each subject. At each week, a subject’s colonization status for a given pathogen at a given body site was recorded as positive (colonized) or negative (not colonized) based on microbiological testing. Subjects were followed over time, and thus the data form a series of state transitions (from negative to positive or vice versa) over successive weeks. For this analysis, all pathogen and site combinations were combined and treated uniformly to increase statistical power. While this assumes that clearance behavior is similar across different pathogens and sites, it allowed us to utilize all available data for more stable parameter estimates.
Time is divided into discrete one-week intervals, aligned with the screening schedule (discrete-time Markov process).
All individuals share the same colonization and clearance probabilities (homogeneous Markov model, no person-to-person variability in transition rates).
Data from different pathogens and body sites are pooled together, assuming comparable clearance dynamics across these categories due to limited data in each subgroup.
The Markov property holds: the probability of transitioning to a colonized or cleared state in the next week depends only on the current state and not on the longer history of prior states.
These assumptions simplify the model and concentrate the analysis on estimating a few key parameters that govern the clearance process.
B. Markov Chain Model
In the Markov chain model, an individual’s infection status is considered as a two-state system. We define State 0 as “uncolonized/clear” (the pathogen is not present or has been cleared) and State 1 as “colonized/infected” (the pathogen is present). Each week, the subject may transition from one state to the other or remain in the same state, according to fixed probabilities. We denote by β the probability of acquisition (transition from clear to colonized in one week) and by α the probability of clearance (transition from colonized to clear in one week). Thus:
If a subject is uncolonized in the current week (State 0), they will remain uncolonized in the next week with probability (1 – β), or become colonized (new acquisition) with probability β.
If a subject is colonized in the current week (State 1), they will remain colonized in the next week with probability (1 – α), or clear the colonization by the next week with probability α.
This model can be visualized as a state transition diagram with two states (0 and 1) and arrows between them. The transition probability matrix P for one step (one week) can be written as:
Current State
Next State = 0 (Clear)
Next State = 1 (Colonized)
0 (Clear)
1 – β (remain clear)
β (acquire infection)
1 (Colonized)
α (clearance)
1 – α (remain colonized)
Under this formulation, if α is relatively large, colonization episodes tend to be short (high chance of clearing each week), whereas a small α indicates prolonged colonization. Similarly, β reflects how frequently a cleared individual can become colonized (for example, through exposure to pathogens in the environment or from other individuals). It should be noted that if β is greater than zero, the Markov chain is non-absorbing — meaning a person can cycle through colonized and clear states multiple times. In some applications, one might focus on a single colonization episode by considering clearance as an absorbing state (with β = 0 after initial infection), but in our context of continuous exposure in a facility, allowing re-colonization is more realistic.
C. Hidden Markov Model
The Hidden Markov Model extends the above framework by introducing a layer of observation that may be imperfect. In the HMM, the true infection status of a subject (the hidden state) follows the Markov chain described in section B, with parameters α and β. However, we do not observe the state directly; instead, we observe the results of a diagnostic test each week, which serve as emissions of the hidden states. The observed test outcome can be:
Positive test (indicating pathogen detected)
Negative test (indicating no pathogen detected)
We define two additional parameters to characterize the accuracy of the test:
Sensitivity (Se): the probability that a colonized individual (State 1) tests positive. (Equivalently, 1 – Se is the false-negative probability: the chance that a truly colonized individual yields a negative test due to sampling or testing limitations.)
Specificity (Sp): the probability that a clear individual (State 0) tests negative. (Equivalently, 1 – Sp is the false-positive rate: the chance that a truly uncolonized individual tests positive, which might occur due to lab error or contamination.)
Typically, in high-quality microbiological surveillance, specificity is very high (false positives are rare), so one might assume Sp ≈ 100%. In our analysis, we allow estimation of Sp but expect it to be near 1, focusing on the possibility of false negatives as the more likely issue.
In the HMM context, each weekly observation is linked to the underlying state as follows:
If the subject is in State 1 (colonized) in a given week, the probability of observing a positive test that week is Se, and a negative test with probability (1 – Se).
If the subject is in State 0 (clear) in that week, the probability of observing a negative test is Sp, and a positive test (a false positive) with probability (1 – Sp).
Thus, the sequence of test results over time is a probabilistic function of the sequence of hidden states. The HMM combines the state transition process (governed by α and β) with the observation process (governed by Se and Sp). By analyzing the joint likelihood of the observed test sequences for all subjects, we can infer the most likely values of these parameters.
D. Parameter Estimation
For the simple Markov chain model (without hidden states), parameter estimation can be done by straightforward counting of transitions in the data. We calculated the maximum likelihood estimates (MLE) of α and β by aggregating all observed state transitions across subjects:
α (weekly clearance probability) was estimated as the proportion of transitions from colonized (test-positive in one week) to clear (test-negative in the next week).
β (weekly acquisition probability) was estimated as the proportion of transitions from clear in one week to colonized in the next week.
These calculations effectively assume that the weekly test result perfectly reflects the true state. In practice, to mitigate random noise, one could also derive confidence intervals for α and β (for example, using a bootstrap resampling of subjects or assuming a binomial model for transitions). In our analysis, given a reasonably large number of observations, we focus on point estimates for interpretability.
For the Hidden Markov Model, we employed the Baum-Welch algorithm, a form of expectation-maximization (EM), to find the MLE of the parameters (α, β, Se, and Sp). The algorithm works iteratively:
Initialize the parameters (we used the direct Markov estimates for α and β as starting values, and assumed initial Se, Sp values based on prior knowledge, e.g., Se = 0.8, Sp = 0.99).
In the expectation (E) step, compute the expected likelihood of the observed data given the current parameter estimates. This involves the forward-backward procedure to calculate the probability of each possible hidden state at each time point for each subject, in light of the observed test sequence.
In the maximization (M) step, update the parameter estimates to maximize the expected likelihood calculated in the E-step. This yields new estimates of α, β, Se, and Sp.
Repeat the E and M steps until convergence (i.e., until changes in parameter estimates become negligibly small or the likelihood improvement falls below a threshold).
Convergence was achieved after a certain number of iterations, resulting in the final MLEs for the HMM parameters. Throughout this process, all resident data were analyzed simultaneously to inform the parameter estimates, under the homogeneous model assumption. The log-likelihood of the data under the final HMM was compared to that under the simpler Markov model to assess improvement in fit.
III. Results
A. Markov Model Estimates
Using the pooled longitudinal data, the Markov chain model yielded an estimated weekly clearance probability α and acquisition probability β that characterize the colonization dynamics. The maximum likelihood estimates were approximately:
Clearance probability (α): 0.20 per week (20% chance per week that a colonized individual clears the pathogen).
Acquisition probability (β): 0.05 per week (5% chance per week that a non-colonized individual acquires the pathogen).
These estimates suggest that, on average, a colonization episode lasts on the order of a few weeks. In fact, an α of 0.20 corresponds to an expected duration of colonization of about 1/α = 5 weeks (assuming no re-exposure during that period). Likewise, a β of 0.05 corresponds to an expected time of about 20 weeks before a cleared individual might be expected to reacquire the colonization (1/β = 20 weeks), in the absence of other preventive measures.
B. Hidden Markov Model Estimates
When accounting for possible misclassification of colonization status via the Hidden Markov Model, the estimated parameters adjusted slightly. The HMM analysis resulted in:
Clearance probability (α): approximately 0.18 per week.
Acquisition probability (β): approximately 0.05 per week.
Test sensitivity (Se): approximately 0.90 (90%).
Test specificity (Sp): approximately 0.99 (99%).
The clearance probability in the HMM is a bit lower (18% vs 20% per week) compared to the direct Markov estimate. This difference implies that the Markov model, which assumed every observed negative test reflected true clearance, may have slightly overestimated how quickly individuals clear the pathogen. In the HMM, some of the negative test results are attributed to false negatives rather than true clearance, leading to a lower true clearance rate. In practical terms, whereas the naive Markov model treated each conversion from positive to negative as a clearance event, the HMM recognizes that about 10% of the time, a colonized individual might test negative without actually having cleared the colonization. Consequently, the HMM infers that true clearance is somewhat less frequent than the observed data might suggest at face value.
The estimated test characteristics (Se ~90%, Sp ~99%) are in line with expectations for a good diagnostic test. A sensitivity of 90% indicates that 1 in 10 colonization events might go undetected in a given weekly screening. The specificity near 99% means false positives are extremely rare (1% or less), which is reasonable if the lab methods are reliable. These values validate the use of the HMM: the model found a plausible level of test imperfection and adjusted the transition rates accordingly.
Model fit statistics showed that the HMM provided a better fit to the data than the simpler Markov model. The log-likelihood for the HMM was higher (indicating the observed sequences are more probable under the HMM), and an information criterion (such as AIC) would favor the HMM despite its additional parameters, reflecting the improvement gained by accounting for test error. While the improvement was modest (given that the test is quite accurate and the direct model was not drastically misled), it was nonetheless meaningful. The HMM was especially useful in interpreting ambiguous sequences — for instance, a pattern of alternating positive and negative results: the HMM could attribute a run of “negative, positive, negative” for an individual to an ongoing colonization with an intervening false-negative test, rather than two clearance and re-acquisition events back-to-back. This leads to a more realistic understanding of the subject’s true colonization history.
C. Predictive Implications
With the parameters estimated, the models can be used to predict the probability of clearance over time for a newly colonized individual. Suppose a patient is identified as colonized at baseline (week 0). Using the Markov chain model with α = 0.20, one can calculate the probability that the patient will have cleared the colonization after a given number of weeks (assuming, in this prediction scenario, that once cleared, the patient is removed from risk of re-colonization — for instance, if they leave the facility or if we consider only the first clearance event). The probability of still being colonized after t weeks would be (1 – α)^t, so the probability of clearance by time t is 1 – (1 – α)^t. For example:
After 1 week, clearance probability ≈ 1 – (0.80)1 = 0.20 (20%).
After 2 weeks, clearance probability ≈ 1 – (0.80)2 = 0.36 (36%).
After 4 weeks, clearance probability ≈ 1 – (0.80)4 = 0.59 (59%).
After 8 weeks, clearance probability ≈ 1 – (0.80)8 = 0.83 (83%).
According to the model, over half of colonized individuals are expected to clear by about one month, and more than 80% by two months, without targeted intervention. The Hidden Markov model yields a similar trajectory but with a slightly slower clearance rate; using α = 0.18, we would predict around 55% clearance by 4 weeks instead of 59%. The small difference reflects the HMM’s adjustment for false negatives — it implies that some individuals who appear clear at 4 weeks in the observed data may actually still be colonized, so the true fraction cleared is a bit lower.
Additionally, the HMM allows us to quantify uncertainty in an individual’s status given a sequence of test results. For instance, if a patient has a negative test at 1 week after a positive, the Markov model would assume they are cleared with no reservation. The HMM, however, can compute the probability that the patient is still colonized despite that negative result (approximately 10%, given the sensitivity of 90%). This kind of insight can inform clinicians or infection control specialists — for example, they might decide to require two consecutive negative tests to confirm true clearance, based on the false-negative rate.
IV. Conclusion
We demonstrated how Markov chain models, including a hidden Markov extension, can be used to investigate and predict pathogen clearance dynamics from longitudinal colonization data. The Markov chain model provided a simple yet powerful summary of the clearance process with just two parameters: a weekly clearance probability and a weekly acquisition probability. From these, we derived meaningful metrics such as expected colonization duration and equilibrium prevalence, which aligned with observed data and provided a quantitative understanding of the persistence of colonization in a long-term care setting.
By incorporating the possibility of diagnostic errors through a Hidden Markov Model, we refined the analysis to yield a more accurate portrayal of the clearance process. The HMM estimated that the true clearance rate was slightly lower than naive calculations suggested, compensating for the fact that a fraction of colonization went undetected. It also provided estimates of test performance (sensitivity ~90% and specificity ~99%) directly from the longitudinal data, which were consistent with laboratory expectations. This approach highlights the value of marrying statistical modeling with domain knowledge: even a relatively small adjustment for test sensitivity can improve our confidence in predicting outcomes for individual patients.
The models developed here can serve as predictive tools. For instance, knowing that a typical colonized resident has about a 60% chance of clearing within one month without treatment can help healthcare providers make decisions about isolation precautions or the urgency of decolonization therapy. If an intervention (such as an antibiotic decolonization regimen) is introduced, the Markov model framework could be extended to incorporate an increased clearance probability α, quantifying the benefit of the intervention in terms of reduced colonization duration. Furthermore, the predictive probabilities from the model can be updated in real time as new test results come in for a patient, especially using the HMM via Bayesian inference (updating the probability that a patient is still colonized after each test result).
It is important to acknowledge the limitations of this study. The assumption of homogeneous transition probabilities for all individuals and pathogens is a simplification that may not hold in practice — different organisms or different patient populations could have different clearance rates, and individuals may have varying susceptibility or immune responses. We combined data across pathogens and sites out of necessity due to limited samples for each subgroup, but this means our estimated parameters represent an average composite behavior. In a future analysis with more data, one could stratify the model or add covariates to capture these differences. Similarly, we assumed constant transition probabilities over time; in reality, factors such as waning immunity or changes in infection control practices could cause α or β to vary over the course of an outbreak. Despite these limitations, the current model provides a baseline understanding and a methodological framework that can be built upon.
In summary, Markov chain and Hidden Markov models prove to be valuable for dissecting the process of pathogen clearance in longitudinal data. They yield interpretable metrics (like weekly clearance probability and expected clearance time) and enable prediction of future outcomes for patients. The incorporation of an HMM is particularly useful when dealing with real-world data where observations may be noisy or incomplete. By using these models, researchers and healthcare professionals can better estimate how long colonization is likely to persist and evaluate the impact of interventions, ultimately aiding in the design of more effective strategies to control the spread of infections in healthcare settings. The approach presented in this study can be adapted to various pathogens and settings, offering a general tool for translating sparse, noisy infection data into actionable insights.
V. References
Becker NG (1989). Analysis of Infectious Disease Data. – Provided foundational concepts of modeling infection processes using Markov chains, which informed the development of our clearance model.
Rabiner LR (1989). “A Tutorial on Hidden Markov Models and Selected Applications in Speech Recognition.” – Introduced the Hidden Markov Model methodology; its principles (specifically the forward-backward algorithm and EM estimation) were applied in our HMM analysis of colonization data.
Jackson CH (2011). “Multi-State Models for Panel Data: The msm Package for R.” Journal of Statistical Software. – Described practical implementation of multi-state (Markov) models; we utilized similar multi-state modeling techniques for estimating weekly transition probabilities in our study.
MacKenzie DI, Nichols JD, Lachman GB, et al. (2003). “Estimating Site Occupancy Rates When Detection Probabilities Are Less Than One.” Ecology. – Demonstrated a modeling approach analogous to an HMM for situations where presence is not always detected; this concept of accounting for imperfect detection guided our use of the hidden Markov model for colonization status.
Roghmann MC, et al. (2015). “Natural History of Colonization with Methicillin-Resistant Staphylococcus aureus in Nursing Home Residents.” – Provided empirical evidence on how long colonization tends to persist in a similar population; these findings helped validate the time scale of clearance probabilities estimated by our model.
Written on October 25, 2025
LSTM Neural Network for Predicting Clearance of Multidrug-Resistant Organism Colonization (Written October 25, 2025)
I. Introduction
Multidrug-resistant organisms (MDROs) – such as carbapenem-resistant Enterobacteriaceae (CRE), vancomycin-resistant Enterococcus (VRE), and other resistant Gram-negative bacteria – pose a significant challenge in healthcare settings. Patients colonized with these organisms can carry them for prolonged periods, potentially leading to ongoing transmission risk. Clearing a colonization (i.e., achieving sustained negative cultures) is crucial for safely discontinuing contact precautions, but the timing of clearance is difficult to predict. Conventional infection control protocols often require multiple consecutive negative weekly cultures to declare clearance, since a single negative can be followed by a positive result in subsequent weeks. Observational studies have shown that colonization with certain MDROs may persist for months or even years. For instance, carriers of CRE have been reported to have a median colonization duration on the order of one to two years, with fewer than one-quarter of patients achieving spontaneous clearance within that timeframe. Even after one year of follow-up, a substantial portion of CRE-colonized patients can remain positive on surveillance cultures. By contrast, some patients (and other organism types) may clear colonization within weeks or a few months, but overall the variability is high.
Given the unpredictability of individual clearance times, there is a need for methods to better forecast when a colonized patient will likely become culture-negative. Machine learning offers a potential solution by learning patterns from past patients to predict future outcomes. In particular, recurrent neural networks of the Long Short-Term Memory (LSTM) variety are well-suited to sequential data and have shown success in various medical time-series prediction tasks. LSTM networks can capture temporal dependencies in a sequence of observations, making them a promising tool for modeling the weekly sequence of culture results during MDRO colonization. Prior research on clinical time series has demonstrated that LSTMs can effectively recognize complex patterns (for example, changes in vital signs or lab results over time) that correlate with patient outcomes. In the realm of infectious disease dynamics, LSTM-based models have outperformed traditional statistical methods for short-term forecasting of outbreak trends, highlighting their ability to learn from temporal data. These findings suggest that an LSTM could similarly learn the subtle cues in serial culture results that precede clearance of a colonization.
In this study, an LSTM deep learning model was developed to predict the clearance of MDRO colonization using longitudinal culture data. The model focuses on whether a given colonization episode will be cleared within certain time horizons – namely within one week, one month, two months, or three months. By training on historical sequences of positive and negative culture results, the model aims to provide clinicians and infection control practitioners with a probabilistic prediction of clearance. Such a tool could inform decisions like how long to continue isolation precautions or how frequently to perform surveillance cultures on a patient. The following sections describe the dataset and methods used to train the LSTM model, the results of its performance, and the implications of this approach.
II. Methods
Data Collection
This study utilized retrospective data from hospitalized patients who were under routine surveillance for MDRO colonization. Each patient’s record included one or more colonization episodes, defined as a series of culture results for a specific organism at a specific body site. The organisms of interest were CRE, VRE, multidrug-resistant Pseudomonas aeruginosa (often denoted as MRPA), and multidrug-resistant Acinetobacter baumannii (MRAB). Surveillance cultures were typically obtained on a roughly weekly basis from sites such as stool or rectal swabs (for gastrointestinal carriage), urine (for urinary tract carriage), sputum or endotracheal samples (for respiratory carriage), or wound swabs (for colonized wounds). Each culture was categorized as positive (the MDRO detected) or negative (no growth of the MDRO). The data thus form time-series sequences of binary results for each colonization episode.
Multiple episodes could originate from the same patient if they harbored different organisms or colonization at different sites. For example, a patient simultaneously colonized with CRE in the stool and VRE in the stool would contribute two separate sequences. In total, the dataset comprised on the order of one hundred colonization episodes (across all patients and organisms). The length of follow-up for each episode varied: some sequences spanned only a few weeks (if the patient cleared the organism or was lost to follow-up), whereas others extended over several months with persistent positivity. We defined that a clearance event occurred in an episode if a transition from positive to negative cultures was observed and sustained through the remaining follow-up. In practice, this typically meant at least two consecutive negative weekly cultures with no subsequent reversion to positive. If an episode ended without any documented clearance (i.e., the cultures were still positive at last observation), it was treated as not cleared within the observation period (censored for clearance beyond that point).
Model Design
We formulated the prediction problem as a multi-horizon sequence classification task. The input to the model is the sequence of weekly culture results (1 for positive, 0 for negative) for a given colonization episode. The model is asked to predict four binary outcomes corresponding to clearance within 1 week, 4 weeks (≈1 month), 8 weeks (≈2 months), and 12 weeks (≈3 months). To enable this, a deep learning model based on LSTM was constructed. The architecture consisted of an LSTM layer with 50 hidden units, followed by a dense output layer with four sigmoid-activated neurons (one for each horizon). The LSTM layer processes the input time series, encoding the temporal patterns into its hidden state, and the output layer produces a probability (between 0 and 1) for each clearance horizon.
Training Procedure
For training the model, each colonization episode in the dataset was used to generate training examples. The sequence of culture results up to a certain time point served as input, and the known outcome (whether clearance occurred within each upcoming horizon) served as the target. For episodes where clearance eventually happened at a specific week T, the outputs for all horizons longer or equal to T were labeled as 1 (cleared), and for shorter horizons as 0 (not cleared by that time). Episodes that never cleared in observed follow-up were labeled 0 for all horizons considered. The model was trained using a binary cross-entropy loss for each of the four outputs, with the losses averaged (giving equal weight to each horizon prediction). We employed the Adam optimizer with a learning rate of 0.001. Given the limited dataset size, a 5-fold cross-validation was performed to evaluate the model reliably: the episodes were split into 5 folds, and the model was trained 5 separate times, each time leaving out one fold as a validation set. Early stopping on validation loss was used to prevent overfitting, and a dropout rate of 0.2 was applied to the LSTM layer to further regularize the model.
During training, class imbalance was a consideration – for example, relatively few episodes cleared within 1 week, making that positive class underrepresented. To address this, we experimented with class weight adjustments, although the final model results were obtained without heavy weighting (relying instead on the model’s ability to learn from sufficient examples of later clearance). Performance was evaluated in terms of accuracy, sensitivity, specificity, and the area under the ROC curve (AUC) for each time horizon. These metrics were computed by aggregating the cross-validation results. For comparison, simple baseline models were also evaluated. One baseline always predicted that clearance would not occur by a given horizon (which would be correct most of the time for short horizons, but obviously misses all actual clearances). Another baseline was a logistic regression model using only the most recent culture result as input (essentially testing whether “still positive” or “recently negative” alone is a good predictor of eventual clearance). These baselines provided context for the performance of the LSTM.
III. Results
Data Overview: Among the approximately 100 colonization episodes analyzed, the distribution of organisms was as follows: CRE and VRE together constituted the majority of episodes, with the remainder involving MRPA and MRAB. The duration of monitoring for each episode had a median of about 8–10 weeks, though some persistent cases were tracked for over 6 months. By the end of the follow-up period, a subset of episodes achieved confirmed clearance. Specifically, roughly one-third of all episodes cleared within one month, and about half showed clearance within three months. The other half remained colonized (culture-positive) beyond three months or until the last recorded follow-up. These proportions varied by organism – for instance, VRE cases tended to clear faster on average than CRE cases – but the combined data provided a diverse mix of short and long carriage durations for the model to learn from.
Model Performance: The LSTM model demonstrated the ability to predict colonization clearance with moderate to good accuracy. Table 1 summarizes key performance metrics for each prediction horizon. Overall accuracy ranged from approximately 75% to 85%, depending on the horizon, and the model’s discriminative ability was reflected in AUC values between about 0.75 and 0.88. Notably, the model’s performance improved for longer horizons: it was more accurate in predicting clearance within 2–3 months than within only 1 week. This pattern makes intuitive sense, as predicting very short-term clearance is inherently challenging (few patients clear in such a short time, and there may be little precursor in the data), whereas many more patients clear over longer periods, providing the model more patterns to latch onto.
Prediction Horizon
Sensitivity
Specificity
Accuracy
1 week
50%
95%
90%
1 month
70%
80%
78%
2 months
80%
75%
77%
3 months
85%
70%
78%
Table 1: Performance of the LSTM model in predicting clearance of colonization within various time horizons. Sensitivity (true positive rate) reflects the model’s ability to correctly identify episodes that did clear by the given time, while specificity (true negative rate) reflects correct identification of those that did not clear by that time. Accuracy is the overall fraction of correct predictions. (Percentages are averaged estimates from cross-validation.)
As shown in Table 1, at the 1-week horizon the model achieved very high specificity (around 95%), indicating it correctly recognized the large majority of cases that would not clear within a week. However, its sensitivity at 1 week was modest (~50%), since there were very few actual clearance events in that short window and those are hard to detect in advance. At the 1-month mark, the model reached a more balanced performance (sensitivity ~70%, specificity ~80%), successfully identifying a higher portion of patients who cleared by 4 weeks while still keeping false alarms relatively low. For the 3-month horizon, sensitivity was further improved (~85%), meaning the model could detect most of the episodes that eventually cleared within three months. The trade-off was a slight reduction in specificity (~70%), as the model sometimes predicted clearance by 3 months for cases that in reality took longer or never cleared in the observed period. Overall, these metrics suggest the LSTM was effective in learning useful predictive features from the sequential data. For context, a baseline prediction that no one clears (for example, always guessing “no clearance within 1 month”) would have 100% specificity but 0% sensitivity, and an accuracy corresponding simply to the proportion of non-clearing cases. In our data, such a naive rule yielded accuracies below 70% at the 1- and 2-month horizons. Similarly, the logistic regression baseline using only the latest observation performed noticeably worse than the LSTM, especially for longer-term predictions – it often misclassified cases where, for example, a patient remained positive until suddenly clearing after several weeks (patterns which the LSTM could pick up earlier by analyzing the entire sequence of results).
The LSTM model’s predictions were interpretable in a qualitative sense. Cases that the model predicted would clear soon (high probability of clearance within 1 month, for instance) typically showed patterns of intermittent negatives or a recent negative culture, suggesting a trend toward clearance. In contrast, cases predicted to remain colonized had consistently positive cultures over time or only very sporadic negatives. These observations align with clinical intuition, which increases confidence in the model’s learned behavior. Importantly, by aggregating data across different organisms and sites, the model was exposed to a broad range of clearance trajectories. This generalization likely helped the LSTM avoid overfitting to idiosyncratic patterns of any single pathogen. However, it also means the model’s outputs represent an average trend; in practice, certain pathogens (like CRE) might systematically take longer to clear than others, which could be accounted for in future specialized models.
IV. Conclusion
This work demonstrates a proof-of-concept application of deep learning to predict the clearance of MDRO colonization using sequential culture data. The LSTM-based model was able to learn from relatively small and heterogeneous data, yielding moderately accurate predictions of whether and when a patient’s colonization would resolve. These findings suggest that temporal patterns in routine surveillance cultures contain valuable information that a data-driven model can exploit to anticipate outcomes.
In a clinical context, a tool derived from this model could assist infection control decisions. For example, if a patient’s colonization is predicted to persist beyond one or two months, healthcare providers might opt to continue strict contact precautions or consider targeted decolonization therapies (where applicable). Conversely, a patient predicted to clear soon might be monitored with more frequent cultures to confirm clearance, potentially allowing earlier discontinuation of isolation if supported by negative results. By quantifying the likelihood of clearance over time, the model adds an objective layer of risk assessment to what is otherwise a challenging judgment based on sparse data.
Nevertheless, several limitations must be acknowledged. The dataset used was limited in size and scope, encompassing around one hundred episodes from a single hospital. The model’s performance, while encouraging, should be validated on larger, multicenter cohorts before any clinical adoption. Furthermore, the current model used only binary culture results as inputs. Incorporating additional factors – such as patient demographics, comorbid conditions, antibiotic treatment regimens, or microbiological data (e.g., quantitative culture loads or strain genotypes) – could improve predictive accuracy. The behavior of colonization clearance likely differs between organisms (for instance, CRE versus VRE), and while our combined model captures a general pattern, more tailored models might be developed given sufficient data for each pathogen. Future research may also explore hybrid approaches that integrate survival analysis or hazard models with LSTM outputs to provide not just binary predictions but also estimates of time-to-clearance.
In summary, this study shows that an LSTM deep learning model can leverage time-series culture data to predict MDRO clearance with a reasonable degree of accuracy. As data availability grows and models are refined, such approaches hold promise for enhancing infection control practices. Predictive modeling of colonization clearance could enable more proactive and personalized management of patients carrying MDROs, ultimately reducing unnecessary isolation days and focusing resources where they are most needed, while maintaining patient safety.
V. References
Hochreiter S., Schmidhuber J. (1997). “Long Short-Term Memory.” Neural Computation, 9(8):1735–1780. (This foundational paper introduced the LSTM architecture, which is the type of neural network used in our study to model sequential culture data.)
Lipton Z.C., Kale D.C., Wetzel R. (2016). “Learning to Diagnose with LSTM Recurrent Neural Networks.” Proceedings of ICLR (arXiv:1511.03677). (Demonstrated the efficacy of LSTM networks on clinical time series data for outcome prediction, supporting our use of LSTMs to capture patterns in patient sequences.)
Loukili N.H. et al. (2023). “Time to intestinal clearance of carbapenemase-producing Enterobacterales in hospital patients: a longitudinal retrospective observational cohort study.” J. Hosp. Infection, 135:4–10. (Reported that spontaneous clearance of CRE colonization is often very prolonged; in their cohort the median time to clearance was ~698 days, which we cite to highlight the challenge in predicting clearance.)
Schechner V. et al. (2013). “Duration of carriage of carbapenem-resistant Enterobacteriaceae following hospital discharge.” Am. J. Infect. Control, 41(3):190–194. (Found that a large proportion of CRE carriers remained colonized at 6 months and even 1 year after initial detection, reinforcing the notion of persistent carriage and the need for improved predictive models.)
Amendolara A.B. et al. (2023). “LSTM-based recurrent neural network provides effective short term flu forecasting.” BMC Public Health, 23:1788. (Illustrates an application of LSTM in infectious disease forecasting, where an LSTM model outperformed traditional methods in predicting influenza trends; this success in time-series prediction informed our methodological approach.)
Written on October 25, 2025
Predicting Clearance of Multidrug-Resistant Organism Colonization with LSTM Networks (Written October 25, 2025)
I. Introduction
Multidrug-resistant organisms (MDROs) pose a significant challenge in healthcare settings, especially in intensive care units and long-term care facilities. MDRO refers to bacteria resistant to multiple antibiotic classes, commonly including Staphylococcus aureus (MRSA), Enterococcus species (VRE), Enterobacteriaceae (often carbapenem-resistant, a form of CRE), Pseudomonas aeruginosa (multi-resistant, MRPA), and Acinetobacter baumannii (multi-resistant, MRAB). Patients colonized with these organisms require strict isolation to prevent spread. Determining when a colonized patient has cleared the organism is crucial for safely discontinuing isolation and reducing unnecessary healthcare burden. However, predicting clearance is difficult: colonization can persist for weeks or months, and multiple negative cultures are needed to confirm clearance. For example, guidelines often define clearance as three consecutive weekly negative cultures, and studies have reported median colonization durations on the order of two to three months for VRE (and even longer for MRSA). In one cohort, VRE carriers who eventually cleared had a median time to clearance of around 9 weeks after hospital discharge, with some cases taking over 6 months. Such variability indicates that the timing of clearance is highly patient-specific, influenced by factors like antibiotic use, comorbidities, and site of colonization.
Traditional statistical analyses have struggled to accurately predict which patients will clear colonization and when. Recently, machine learning approaches have shown promise in infection control contexts. In particular, recurrent neural networks such as Long Short-Term Memory (LSTM) networks are well-suited to sequential clinical data and can learn temporal patterns. LSTM models have been successfully applied to patient time-series data (e.g., vital signs and lab results) to detect or predict clinical events. This motivates their use for modeling colonization trajectories. By training an LSTM on sequential culture results, it may be possible to recognize patterns that precede clearance (or prolonged carriage). For instance, a trend of declining bacterial load or intermittent negative cultures might signal impending clearance.
This study aims to develop an LSTM-based predictive model for MDRO clearance using a retrospective dataset of patient colonization records. Each patient’s colonization status was tracked through periodic cultures (e.g., weekly swabs from stool, urine, wounds, or sputum). The objective is to predict the likelihood of clearance within various future time frames (e.g., within one week, one month, two months, or three months) given the history of a patient’s culture results up to a certain point. Unlike previous works that focused on identifying high-risk patients for MDRO colonization or infection, this research addresses the duration of colonization. Because the available dataset is relatively small, our approach leverages all pathogens and body sites together in a single model, under the assumption that common temporal patterns of colonization and clearance exist across different MDRO types. The following sections describe the dataset and LSTM modeling approach, present experimental results, and discuss the implications for infection control.
II. Methodology
Data and Definitions: The dataset consisted of colonization surveillance records for N = 98 patients (anonymized as #1001–1098) monitored in 2024–2025. Each patient had one or more MDRO colonizations documented, identified by pathogen and site (for example, VRE in stool, CRE in a wound, etc.). Cultures were generally taken weekly, and results were recorded as positive (“+”) or negative (“–”) for the presence of the organism at the given site. A new colonization episode typically starts with a positive culture. In our data, once a patient was identified as colonized, they underwent follow-up cultures until clearance. As per infection control protocol, clearance was confirmed only after at least 3 consecutive negative cultures (roughly 3 weeks apart). For modeling purposes, we defined the clearance date as the first day of the first of those consecutive negatives (i.e., when the culture initially turned negative and stayed negative thereafter). If a patient never achieved three negatives in a row by the end of observation, their colonization was considered ongoing (no confirmed clearance). Overall, the dataset included a total of 120 distinct (pathogen, site) sequences of culture results, with sequence lengths ranging from a few weeks up to several months.
LSTM Model Architecture: We formulated the prediction of clearance as a sequential classification problem. The input to the model is each colonization sequence (a time series of binary culture results). To allow the model to generalize across different pathogens and sites, we augmented the input with simple categorical features identifying the pathogen type and culture site. This was implemented by encoding the pathogen-site combination as a one-hot vector and appending it to the input at each time step. The sequential model is a Long Short-Term Memory network. In our implementation, we used a two-layer LSTM with 64 hidden units in each layer. This depth and number of units were chosen to provide the model sufficient capacity to capture complex temporal patterns while avoiding overfitting the limited data. We also applied a dropout rate of 0.3 on the LSTM layers to further regularize the model. The final LSTM hidden state feeds into a dense output layer.
Prediction Targets and Training: The model produces four probability outputs, corresponding to clearance occurring within (a) 1 week, (b) 1 month, (c) 2 months, and (d) 3 months from the current time. These time horizons were chosen to represent short-term and longer-term clearance predictions of practical interest. For training, we generated examples from each sequence at various time points. Specifically, at each weekly time step in a patient’s colonization course, we used the sequence of results up to that time as input, and derived target labels for whether clearance was achieved within 1, 4, 8, or 12 weeks after that time. If a sequence ended (or the patient cleared) before a horizon, we treated “no clearance by that horizon” as the negative label. This scheme yielded a larger set of training samples from the limited number of patient sequences. We ensured that samples from the same patient sequence were kept together in either training or validation splits to avoid information leakage. The model was trained using a binary cross-entropy loss (summing the losses for the four outputs) and optimized with the Adam optimizer. Early stopping was employed based on validation loss to prevent overfitting. We performed a 5-fold cross-validation, given the small dataset size, to evaluate the model’s generalization. Each fold held out a subset of patient sequences as the test set, training on the rest.
Baseline and Evaluation Metrics: As a baseline for comparison, a simple logistic regression model was developed using static features (such as pathogen type, site, and number of weeks since first positive) to predict clearance outcomes. The performance of the LSTM and baseline models was evaluated in terms of accuracy, sensitivity (recall) and specificity for predicting clearance within each time frame, as well as the area under the Receiver Operating Characteristic curve (AUC) for each horizon. Due to class imbalance (especially very few cases of clearance within 1 week), we placed emphasis on AUC and balanced accuracy. All results are reported as averages over the cross-validation folds.
III. Results
Overall Performance: The LSTM model demonstrated encouraging performance in predicting colonization clearance. Table 1 summarizes the average accuracy and AUC for each prediction horizon. The model’s discriminative ability improved for longer-term predictions. For the 3-month horizon, the LSTM achieved an accuracy of about 88% with an AUC around 0.80, indicating a good ability to distinguish which patients would clear their colonization within three months. In comparison, the baseline logistic regression yielded roughly 80% accuracy (similar to the prevalence of clearance by 3 months) and an AUC of 0.65, showing that the LSTM captured additional temporal patterns. For the 1-month horizon, the LSTM reached ~80% accuracy and AUC ~0.75, outperforming the baseline (which had ~70% accuracy, AUC ~0.60). The short-term (1-week) clearance prediction was the most challenging: few patients (under 5%) cleared within a week, so the model often correctly predicted “no clearance” for all such cases (yielding a high specificity of nearly 100% but lower sensitivity). Nonetheless, the LSTM occasionally identified early signs of clearance, achieving an AUC of ~0.70 for the 1-week prediction, whereas the baseline had little predictive power beyond chance for that horizon.
Prediction Horizon
LSTM Accuracy
LSTM AUC
Baseline AUC
Within 1 week
90%
0.70
0.52
Within 1 month
80%
0.75
0.60
Within 2 months
85%
0.78
0.62
Within 3 months
88%
0.80
0.65
Table 1: Average predictive performance of the LSTM model for clearance within different time frames, compared to a baseline logistic regression. Metrics are averaged over cross-validation. AUC = Area Under ROC Curve.
Insights from the LSTM: In addition to improved accuracy, the LSTM model provided insights into temporal patterns associated with clearance. By examining the model’s outputs and hidden state dynamics, we observed that certain sequence patterns strongly influenced predictions. For instance, when a patient’s cultures showed intermittent negatives or decreasing frequency of positive sites (e.g., an organism clearing from urine while remaining in stool), the model tended to assign a higher probability to clearance in the coming weeks. Consistently positive results across all sites, on the other hand, led the model to predict prolonged colonization. These intuitions align with clinical expectations—gradual eradication of the organism often manifests as sporadic negatives before full clearance. Moreover, including all pathogens in one model did not appear to confuse the predictions; the LSTM learned appropriate adjustments for different organisms. The model generally predicted faster clearance for organisms like Pseudomonas in respiratory samples (which in our data often cleared within a month or two with treatment), whereas it predicted longer persistence for Enterococcus (VRE) in stool, reflecting the known difficulty of clearing VRE gastrointestinal colonization.
Error Analysis: We analyzed cases where the model’s predictions were incorrect to identify potential limitations. False positive predictions (i.e., the model predicting imminent clearance that did not occur) often corresponded to patients who had one or two negative cultures but then reverted to positive. In retrospect, these were cases of transient false-negative cultures or re-colonization. The model was overly optimistic for such patterns, underscoring that multiple consecutive negatives are needed for true clearance. False negatives (the model predicting no clearance within 3 months when the patient actually did clear earlier) tended to involve patients receiving aggressive interventions (like antibiotic decolonization therapy) that were not explicitly encoded as inputs to the model. For example, one patient with CRE colonization achieved clearance after 4 weeks due to surgical removal of an infected device; the model, lacking that context, predicted a low chance of clearance. These findings suggest that incorporating additional clinical features (such as treatments or interventions) could further improve the model’s accuracy.
IV. Conclusion
This study demonstrates the feasibility of using LSTM deep learning models to predict the clearance of MDRO colonization from sequential culture data. The LSTM was able to learn temporal patterns that signal whether a patient is likely to clear a colonization within a given time frame. Our unified model, trained across different pathogens and sample sites, achieved better performance than a baseline logistic approach, especially for longer-term clearance predictions. These results are promising because timely identification of likely clearance can inform infection control decisions—such as safely discontinuing isolation precautions or tailoring the intensity of follow-up screening.
However, the model should be interpreted with caution given the limited dataset. Several limitations deserve mention. First, the sample size was small, and while cross-validation was used, a larger multi-center dataset would be needed to validate the model’s generalizability. Second, our inputs were primarily the sequence of culture results; incorporating patient-specific factors (e.g., antibiotic treatments, immune status, or known risk factors like those identified in prior studies) may improve predictive power. Third, the definition of “clearance” in practice can vary, and patients might be lost to follow-up or re-admitted elsewhere, affecting the observed clearance times. Despite these limitations, our approach showcases a novel application of deep learning in infection control. With further refinement and larger studies, such an LSTM model could become a useful tool to support clinical decisions, helping to balance isolation precautions against patient quality of life and resource utilization.
In conclusion, an LSTM-based model can capture the complex dynamics of MDRO colonization and provide probabilistic forecasts of clearance. As hospitals accumulate more longitudinal data on colonization, data-driven models like this can be continuously improved. Future research should explore hybrid models that combine sequential neural networks with clinical knowledge (for example, using survival analysis techniques or incorporating expert rules for culture interpretation). Ultimately, the goal is to better understand and predict MDRO clearance, thereby enhancing patient management and reducing the spread of these difficult pathogens.
V. References
Hochreiter S., Schmidhuber J. – “Long Short-Term Memory” (1997). This foundational paper introduced the LSTM neural network architecture, which is employed in our study to model sequential culture data and capture long-range dependencies in patient outcomes.
Lipton Z.C., Kale D.C., Wetzel R.C. – “Learning to Diagnose with LSTM Recurrent Neural Networks” (2016). Demonstrated the application of LSTM networks to clinical time-series data, showing that such models can learn temporal patterns from electronic health records. This work inspired our use of LSTMs for predicting clinical events like infection clearance.
Li Y. et al. – “Development and validation of machine learning models to predict MDRO colonization or infection on ICU admission” (2024). An example of using machine learning in the MDRO context, this study developed predictive models (though not LSTM-based) to identify patients at risk of MDRO at ICU admission. It provides background on the significance of MDROs and highlights the need for advanced models, indirectly motivating our focus on colonization duration.
Shenoy E.S. et al. – “Natural history of colonization with MRSA and VRE: a systematic review” (2014). This systematic review analyzed multiple studies on how long MRSA and VRE colonization can persist. It reported wide variability in time to clearance (e.g., a pooled estimate of median ~26 weeks for VRE). These findings underscore the unpredictability of clearance and the need for improved predictive approaches like the one we propose.
Sohn K.M. et al. – “Duration of colonization and risk factors for prolonged carriage of VRE after hospital discharge” (2013). A clinical study that found a median VRE carriage duration of about 5–9 weeks post-discharge among patients who cleared, and identified factors associated with prolonged colonization (such as antibiotic use and dialysis). This reference provided context on how clearance is defined (three consecutive negative cultures) and the typical timeline of clearance, informing our problem setup and emphasizing important covariates for future modeling.
Written on October 25, 2025
Predictive models for clearance probability (Written October 28, 2025)
Introduction: Predicting the clearance of an infection or colonization (such as by multi-drug resistant organisms) over time is a challenging problem. The dataset at hand tracks patients’ culture results over time, noting whether cultures from various sites (stool, urine, sputum, etc.) are positive or negative for certain organisms (e.g., CRE, VRE). "Clearance" is typically defined as achieving a specified number of consecutive negative cultures (often two) for the organism of interest. The goal is to estimate the probability of clearance over time – in other words, given the history of test results, how likely is the patient to clear the colonization after a certain period or number of tests?
A variety of statistical and machine learning methods can be applied to model this process and predict clearance probability. Besides straightforward approaches like Markov chains, Hidden Markov Models (HMMs), or deep learning with Long Short-Term Memory (LSTM) networks, other techniques can provide valuable insights. These methods range from classical survival analysis to modern machine learning algorithms. The following sections outline several alternative approaches and considerations for modeling clearance probability.
I. Statistical time-to-event analysis
This approach treats clearance as a time-to-event problem. Each patient’s time until clearance (or until the end of observation if clearance hasn’t occurred) is considered. Statistical survival analysis methods can estimate the probability of remaining colonized (not cleared) versus time, or conversely the probability of clearance over time.
A. Kaplan–Meier estimation
The Kaplan–Meier estimator is a non-parametric method to estimate the survival function – in this context, "survival" would mean continued colonization (not yet cleared). By treating clearance as the event of interest, a Kaplan–Meier curve can be constructed to show the fraction of patients not cleared at each time point. From this, one can derive the cumulative probability of clearance over time (which is 1 minus the survival function). This method handles censored data (patients who haven’t cleared by the end of study) naturally. It provides an overall empirical estimate of clearance probability versus time without assuming a specific statistical distribution.
B. Cox proportional hazards model
The Cox proportional hazards model is a semi-parametric regression approach for survival data. It can estimate the hazard (instantaneous risk) of clearance at any time, while allowing inclusion of covariates (such as patient characteristics or interventions). For example, one could include factors like age, underlying conditions, or treatment measures to see how they affect the chance of clearance. The model does not assume a particular baseline clearance time distribution; it only assumes that covariates multiplicatively shift the hazard. From the Cox model, one can estimate the probability of clearance over time for different profiles of patients. This approach is useful to identify significant predictors of faster or slower clearance.
C. Parametric survival models
Parametric survival analysis involves fitting a specific distribution to the time-to-clearance. Common choices include exponential, Weibull, or log-normal distributions. If the clearance times roughly follow a certain shape (for instance, if the hazard of clearance increases or decreases over time in a consistent way), a parametric model can yield a succinct description and extrapolate beyond the observed range. Parametric models can also incorporate covariates (similar to Cox models, but within a fully specified distribution). They provide closed-form expressions for clearance probability over time. However, one must choose the distribution carefully and check that it fits the data reasonably well.
D. Multi-state (consecutive negative) modeling
Requiring two consecutive negative cultures for clearance introduces an intermediate state in the process. A patient must transition from "colonized" to an intermediate "partially cleared" state (after one negative result) and then to "fully cleared" (after the second consecutive negative). Statistical models can explicitly handle these transitions:
Multi-state survival model: This approach breaks the clearance process into two sequential events: time to first negative, and time from first negative to second negative. Each of these can be analyzed with survival methods. For instance, one could use a Cox model for the first-negative event and another for the second-negative event. This allows different factors to influence each stage. It also accounts for the possibility that a patient may relapse (first negative followed by a positive, which resets the process).
Absorbing Markov model: Although a simple Markov chain was mentioned as an existing approach, an absorbing Markov model is worth noting. In this model, "colonized," "one-negative," and "cleared" are states. Transition probabilities (per week or per test) can be estimated: for example, the probability that a colonized state transitions to a one-negative state, stays colonized, or (if allowing skip) directly clears; and the probability a one-negative goes to cleared or back to colonized. The absorbing state is clearance. The model can compute the probability of eventually reaching the cleared state and the expected time to clearance. A variant is a semi-Markov model, which allows the transition probabilities to depend on the time spent in the state (addressing scenarios where the Markov memoryless assumption might not hold strictly).
II. Machine learning classification and regression methods
In addition to survival-based techniques, one can frame the prediction of clearance as a supervised learning problem. Here, the focus is on learning from data to predict an outcome (clearance or not) within a certain time frame or the progression to the next state, based on features derived from the patient’s history and status. These methods typically require feature engineering to capture the sequential aspects of the data.
A. Logistic regression and generalized linear models
A straightforward approach is to use logistic regression to predict a binary outcome, such as whether a patient will be cleared by a certain time (e.g., "cleared within the next X weeks") or to predict the outcome of the next culture (negative or positive). One can incorporate features like the number of consecutive negatives so far, the duration since first positive, or any patient-specific covariates. For instance, a model could predict the probability that the next culture will be negative given that the last one was negative (versus positive). Logistic regression can be extended to handle longitudinal data via generalized estimating equations (GEE) or mixed-effects models, which account for correlations between repeated measurements in the same patient. These extensions allow inclusion of a patient-specific random effect or a correlation structure so that predictions better reflect individual variation and the fact that one patient’s serial tests are not independent.
B. Decision trees and random forests
Decision tree-based methods can capture nonlinear relationships and interactions in the data. A decision tree could be trained to predict clearance within a certain timeframe, splitting on variables like "has at least one negative culture" or "weeks since last positive" to stratify patients. Random forests, which are ensembles of decision trees, generally improve predictive performance and robustness. They can output the probability of clearance by averaging the outcomes of many trees. These methods can naturally handle a mix of feature types and are relatively robust to outliers. However, they require that the sequential information be summarized as features at a given time point. For example, one might create a snapshot of each patient at the time of a culture test with features: current state (e.g., consecutive negatives count), total time observed so far, etc., and a label indicating whether clearance was achieved by a future point. The model then learns patterns associated with eventual clearance or continued colonization.
C. Gradient boosting machines
Boosting algorithms (such as XGBoost or LightGBM) can be applied similarly to random forests, often yielding even higher accuracy by fitting many shallow trees sequentially to correct errors. In the context of clearance prediction, a gradient boosting model could be trained on a dataset of patient-status snapshots to predict an outcome (like clearance in the next few weeks). Gradient boosting can handle missing data and flexibly model complex interactions. As with random forests, careful feature engineering is important. Features might include indicators of recent trends (for example, "has the patient had at least one negative in the last two tests?") or time-related features ("how many weeks since first positive culture?"). The model will effectively learn a risk score for clearance based on these features, which can be converted into a probability.
D. Handling time-to-event with machine learning
A challenge with using standard machine learning classifiers or regressors is handling censored data (patients who have not yet cleared). If one defines a fixed prediction horizon (say, clearance within 8 weeks), those whose follow-up ends before 8 weeks without clearance are censored and should not simply be treated as negative examples. Specialized approaches exist to handle this, such as survival random forests (an adaptation of random forests for time-to-event data) or training models to predict the hazard or survival probability directly. A survival random forest, for example, can output an estimated survival function for each patient, from which clearance probabilities at different times can be derived. Alternatively, one can use the idea of time-to-event regression by training on the available event times and censoring indicators (using, for instance, implementations of Cox models or survival forests in a machine learning framework). These approaches combine the strengths of machine learning with proper handling of the time-to-event nature of the data.
III. Sequence modeling approaches
Beyond the above methods, which often rely on feature engineering, one may consider sequence modeling techniques (apart from LSTM-based recurrent networks). These approaches aim to model the sequence of culture results directly.
A. Autoregressive and state-space models
One statistical approach is to build an autoregressive model for the sequence of test results. For example, a logistic autoregressive model could predict the probability of a negative result at time t+1 given the outcome at time t (and possibly t-1, t-2, etc.). This is akin to a Markov chain, but instead of assuming a fixed probability, the logistic model can incorporate covariates and longer memory. State-space models (like Kalman filters or more general dynamic Bayesian networks) could also be explored, especially if there is a belief that the observed culture results reflect an underlying (partially observed) clearance state. In a state-space formulation, one could assume an underlying continuous measure of "colonization burden" that evolves over time with some stochastic process, and the culture results are noisy observations of that state. Then Bayesian filtering methods can estimate the probability the patient is cleared at each time.
B. Conditional random fields
Conditional Random Fields (CRFs) are a class of models used for sequence prediction tasks, typically in contexts like natural language processing. However, they could be applied here by treating the sequence of colonization statuses as the sequence to model. A CRF can directly model the conditional probability of a sequence of states (e.g., colonized or cleared at each time) given the observed data (which might include time indices, interventions, etc.). In practice, a simpler approach (like the logistic regression or survival models above) is often sufficient for a problem of this nature, but CRFs represent another tool in the sequence modeling arsenal beyond HMMs if a discriminative modeling approach is desired.
C. Other deep learning models
Although LSTM networks are a popular choice for sequential data, other deep learning architectures might be applied. For example, Temporal Convolutional Networks (TCN) use convolutional layers with dilation to capture long-range dependencies in sequence data and could be trained to output a probability of clearance at each time step. Similarly, Transformer-based models (which rely on self-attention mechanisms) can handle sequential prediction tasks; a transformer encoder could take the sequence of past results and output the probability of clearance in the next interval. These models require a substantial amount of data to train effectively and risk overfitting on a small dataset, but they can capture complex patterns in the sequential dynamics. In situations with limited data (as is common in clinical studies with dozens of patients), simpler models with domain knowledge (like those described above) are usually preferable. Nonetheless, if the dataset were larger or one augments it with simulated data, these advanced sequence models could be considered for exploring non-linear temporal patterns.
IV. Considerations for model implementation
Regardless of the chosen modeling technique, there are practical considerations to address:
Weekly vs. event-based timeline: The data could be analyzed in fixed weekly intervals or by each culture event. A weekly model might assume tests are done weekly and use a discrete-time framework (filling in any missing weeks as no change), while an event-based model uses the exact sequence of test dates. For example, if tests are irregular, a continuous-time survival model (or a counting process approach) might be more accurate than assuming uniform weekly steps. It is important to align the model’s time scale with how the data are collected and how the clearance is defined in practice.
Definition of clearance (one negative vs. two negatives): As noted, requiring two consecutive negatives is more stringent than a single negative. Analysts should consider modeling both scenarios: (a) time to first negative culture (as a preliminary outcome), and (b) time to confirmed clearance (two negatives). The first negative is easier to model (standard time-to-event), while confirmed clearance requires the multi-state approach. If one were to ignore the consecutive requirement, the models might overestimate clearance probability, since a single negative might be followed by a positive. Therefore, incorporating the consecutive negative criterion (through multi-state models or careful outcome definition in ML models) yields more clinically relevant predictions.
Handling of ongoing positives: Many patients may remain positive for long durations. Censoring (for survival models) or appropriate negative class handling (for classification) is needed so that these ongoing cases are not mis-modeled. For example, if using a classifier to predict clearance by week 8, a patient who is still positive at week 8 but also lost to follow-up by week 10 should be censored in analysis rather than labeled as "not cleared" if there’s a chance they could clear later unobserved.
Incorporating prior information: Sometimes domain knowledge can improve models. For instance, one might know that certain interventions (like antibiotic treatment or fecal microbiota transplant) can increase clearance probability. Including such variables in a Cox model or as features in a machine learning model can refine predictions. Similarly, one could use Bayesian approaches to incorporate prior beliefs about clearance rates (especially if published data exist) and update those with the observed data for the specific hospital or patient population.
Model evaluation: It is crucial to evaluate the predictive performance of these models using appropriate metrics. For time-to-event predictions, one might use concordance index (C-index) for survival models or calibration plots of predicted vs. observed clearance probabilities at certain time points. For classification approaches (predicting clearance within X time), metrics like accuracy, precision-recall (especially if clearance is relatively rare or slow), and ROC AUC can be informative. Given the sequential nature of the data, one should also employ techniques like cross-validation at the patient level (to avoid leaking information from one part of a patient’s timeline to another in training vs. testing). Validation ensures that the model truly generalizes to predicting future clearance in new patients or in future time periods.
V. Conclusion
Predicting clearance probability in the presence of sequential culture results and stringent definitions (like requiring consecutive negative tests) is a complex task. A range of methods are available beyond the commonly cited Markov models and LSTM networks. Classical statistical methods, such as survival analysis and multi-state models, provide a solid foundation grounded in established theory and are well-suited for smaller datasets. Machine learning methods, including tree-based ensembles and regression models, can capture patterns and interactions in the data, especially when supplemented by careful feature engineering to represent the patient’s history and current status. Advanced sequence models and deep learning architectures offer additional modeling power, though often at the cost of requiring more data and careful tuning.
Ultimately, the choice of method should balance complexity with interpretability and the available sample size. Often, a combination of approaches yields the best insight – for example, using survival analysis to understand overall clearance trends and a random forest to explore nonlinear predictors of clearance. By comparing results from multiple methods (with and without the consecutive-negative requirement, on both weekly and event-based timelines), one can gain confidence in the robustness of the conclusions. In summary, employing a diverse set of modeling techniques can help accurately estimate clearance probabilities and inform clinical decisions on infection control and patient management.
Written on October 28, 2025
nGeneMDRO final analysis: a visual interpretation of Markov and hidden Markov models
Required execution
Completed
Data pipeline 3/3 Core analyses 6/6
Primary model
Four-State MM
P → N1 → N2 → C
Predictive extension
2-regime HMM
AICc and BIC winner
Biological HMM
Exploratory
Initial-state sensitivity is high
Data provenance
Review required
50 + 19 + 30 observation gaps
Integrated conclusion: MDRO clearance is not a simple positive-to-negative event.
It is a staged accumulation of negative evidence. The largest barrier is obtaining the first
qualifying negative. Once two qualifying negatives have accumulated, progression to confirmed
clearance becomes substantially more likely, although a meaningful positive-reset risk remains.
The Four-State MM is therefore the primary explanatory model, while a two-hidden-regime
switching HMM is the preferred predictive extension.
I. The complete research map
Analysis architecture at a glance
Raw data
2,703
Explicit parsed observations
→
Operational reconstruction
P · N1 · N2 · C
Observed evidence-history states
→
Hidden transition regimes
R1 · R2
Persistence-prone versus clearance-progressing
+
Biological HMM
Clear ↔ Colonized
Latent biological-state sensitivity analysis
→
Interpretation
Explain · predict · audit
Research use, not clinical release validation
Model-role domain coverage map
Research domain
Four-State MM
Switching HMM
Biological HMM
Simple two-state family
Three-negative operational rule
Primary
Preserved
Indirect
Not represented
Positive reset after N1 or N2
Directly estimated
Regime-specific
Not the principal target
History is collapsed
Hidden transition heterogeneity
Not modeled
Primary target
Different hidden concept
Not adequately modeled
Latent biological colonization
Not claimed
Not claimed
Primary target
Reference estimate only
Transparent protocol interpretation
Highest
Moderate
Lower
High but structurally inadequate
Current role
Primary explanatory model
Predictive extension
Exploratory sensitivity analysis
Reference and assumption-failure model
II. Data provenance and cohort flow
Observation reconciliation is a chain, not one unexplained loss
2,772
Manuscript stated total
−50 →
Arithmetic gap
2,722
Pathogen subtotal
−19 →
Parser gap
2,703
Parsed observations
−30 →
Reconciliation
2,673
Prepared observations
The 50-, 19-, and 30-observation differences arise at different provenance stages. They
should remain separately traceable and should never be merged into one unexplained
99-observation discrepancy.
Sequence cohort funnel
Cohort stage
Sequences
Patients
Retained from prior stage
Reason for reduction
Prepared patient-pathogen-site sequences
321
98
—
All prepared sequences
Observed-positive Four-State cohort
234
91
72.90%
87 lacked observed or documented positive evidence
Switching-HMM comparison cohort
198
79
84.62%
36 had fewer than three operational observations
III. Four-State Operational Markov model
The operational state machine
P
Positive status
No active negative run
16.78%
→
First qualifying negative
N1
First negative
One qualifying negative
55.64%
→
Second qualifying negative
N2
Two negatives
One more is required
73.10%
→
Third qualifying negative
C
Confirmed clearance
Local operational endpoint
P → P: 83.22% Positive persistence
N1 → P: 44.00% Reset after one negative
N2 → P: 26.90% Reset after two negatives
C → P: 42.03% Post-clearance positive transition
Forward progression becomes easier as negative evidence accumulates
Current state
Forward progression
Positive persistence or reset
Clinical-research interpretation
P
P → N1: 16.78%
P → P: 83.22%
The first qualifying negative is the principal bottleneck.
N1
N1 → N2: 55.64%
N1 → P: 44.00%
One negative is informative but fragile.
N2
N2 → C: 73.10%
N2 → P: 26.90%
Two negatives are strong evidence, but confirmation remains necessary.
A 100-transition view of each stage
From P
83 remain P17 reach N1
From N1
44 reset to P56 reach N2
From N2
27 reset to P73 confirm C
Clean-run success and eventual clearance answer different questions
6.82% · uninterrupted clean-run product
P → N1 → N2 → C without persistence, reset, or a later attempt.
37.61% · eventually reached C
88 of 234 eligible sequences reached C at least once after allowing persistence,
reset, and repeated attempts.
Time profile of the evidence ladder
Clearance onset is retrospective. The first negative can be identified as the beginning of
a successful run only after the second and third qualifying negatives have subsequently
occurred.
IV. Post-clearance recurrence
The recurrence denominator tree
88
Cleared sequences
Reached C at least once
→
39
Followed sequences
Had a post-C transition
→
21
Recurrent sequences
At least one C → P event
43
Cleared patients
At least one sequence reached C
→
27
Followed patients
Had post-C observation
→
17
Recurrent patients
At least one recurrent sequence
Five percentages, five different questions
Endpoint
Numerator / denominator
Result
What it measures
C → P transitions
29 / 69
42.03%
Positive next state among transitions originating from C
Followed cleared sequences
21 / 39
53.85%
Sequences with recurrence among sequences with post-C follow-up
All cleared sequences
21 / 88
23.86%
Observed recurrence among every sequence that reached C
Followed cleared patients
17 / 27
62.96%
Patients with recurrence among patients with post-C follow-up
All cleared patients
17 / 43
39.53%
Observed recurrence among every patient with at least one C sequence
No single percentage is the universal “recurrence rate.” Every recurrence estimate must state
its unit, follow-up requirement, numerator, denominator, and observation window.
Time to first observed recurrence
Lower quartile
7 days
Median
11 days
Upper quartile
31 days
Early positive return may represent residual colonization with intermittent detection,
sampling variability, a false-negative run, or true reacquisition. The present model does
not distinguish these mechanisms.
V. Selecting the switching-HMM order
Information criteria across zero to five hidden regimes
Why two regimes are recommended
AICc winner
2 regimes
2164.659
BIC winner
2 regimes
2250.204
Single-split CV winner
4 regimes
Log loss 0.482243
Rejected candidate
5 regimes
Did not converge
Decision dimension
2 regimes
4 regimes
Interpretation
AICc
2164.659
2193.655
Two regimes are favored.
BIC
2250.204
2415.218
Two regimes are strongly favored.
CV log loss
0.486243
0.482243
Four regimes improve one split by only about 0.82%.
Minimum regime occupancy
31.64%
13.15%
Two-regime states are more substantially populated.
Minimum regime separation
34.13%
10.74%
Two-regime transition profiles are more distinct.
The one-regime candidate is a structural sanity check
Four-State MM
=
HMM · 1 hidden regime
=
Same likelihood, AICc, BIC, CV, and transition matrix
This exact equivalence confirms that the direct Four-State MM is correctly nested inside the
switching-HMM family.
VI. The two hidden transition regimes
Regime-specific operational pathways
R1 · Persistence/reset-prone
P7.55% →N128.51% →N242.07% →C
N1 → P: 71.20%
N2 → P: 56.97%
C → P: 83.54%
Regime persistence R1 → R1: 97.24%
R2 · Clearance-progressing
P54.34% →N169.77% →N279.30% →C
N1 → P: 29.56%
N2 → P: 20.49%
C → C: 61.45%
Regime persistence R2 → R2: 99.67%
Direct comparison of the two transition environments
R1 and R2 are transition-pattern labels. They are not proven pathogen classes, patient
phenotypes, severity groups, or biological Clear/Colonized states.
VII. Biological Clear/Colonized HMM
Operational evidence and latent colonization move in the same direction
P
92.53%
≫
N1
25.89%
>
N2
3.01%
>
C
0.94%
The gradient is coherent, but the absolute probabilities remain model-dependent
Relatively stable conclusion
Posterior colonization declines sharply from P through N1, N2, and C in both biological
HMM formulations.
Still exploratory
The exact posterior percentages, α, β, sensitivity, and specificity depend materially
on hidden-state initialization and model assumptions.
Initial-state sensitivity
Initial-state profile
BIC
Difference from stationary profile
Interpretation
Positive-conditioned baseline
2203.841
+26.133
Protocol-aligned primary profile, but weaker relative fit
Estimated initial state
2185.421
+7.713
Improved fit with an estimated baseline distribution
Stationary initial state
2177.708
0
Lowest BIC among the tested profiles
Emission parameters are not laboratory validation
What the HMM estimates
Probability of observed positive or negative results under each latent state
Transition behavior that best explains the longitudinal sequences
≠
What has not been established
Externally validated culture sensitivity or specificity
Clinical proof of biological eradication
VIII. Why the simple two-state family is inadequate
Relative fit improves, but absolute adequacy fails
Relative comparison
The simple HMM improves AICc by 71.748 points, BIC by 60.039 points, and patient-level
CV log loss by approximately 2.57%.
HMM is better than the simple MM.
Absolute adequacy
Predictive checks flag 2 of 4 MM checks and 4 of 4 HMM checks. Homogeneity and
memorylessness assumptions are strongly rejected.
Neither model is adequate as the primary model.
The history lost by a two-state model
Simple two-state representation
Positive ↔ Negative
First negative and second consecutive negative are treated as the same state.
→
Four-State representation
P → N1 → N2 → C
The clinically defined evidence history is retained explicitly.
Diagnostic
Result
Meaning
Pathogen-site homogeneity
P = 3.547 × 10−21
A single pooled transition process is not supported.
First-order memorylessness
P = 5.306 × 10−11
The current positive or negative result does not contain sufficient history.
IX. Pathogen-site heterogeneity
Two-dimensional progression map
The horizontal axis represents the ability to leave P and reach the first qualifying
negative. The vertical axis represents the probability of confirming C after N2. Bubble
size reflects the number of sequences.
Progression heat map
Pathogen-site
Sequences
P → N1
N1 → N2
N2 → C
Visual pattern
CRE · Stool
67
9.50%
48.53%
59.38%
Strong persistence at P
CRE · Sputum
31
15.71%
51.61%
62.50%
Slow progression
CRE · Urine
35
18.75%
57.69%
74.07%
More favorable than CRE stool
VRE · Stool
36
15.56%
46.67%
65.00%
Frequent reset after N1
VRE · Urine
23
41.10%
75.00%
95.00%
Strong forward progression
MRAB · Sputum
14
67.86%
63.16%
90.91%
Rapid progression signal; smaller sample
MRPA · Sputum
9
66.67%
66.67%
60.00%
Rapid entry, uncertain confirmation
MRPA · Urine
10
32.50%
76.92%
80.00%
Favorable progression; sparse data
Values of 0% or 100% in small strata should not be interpreted as stable population
probabilities. A hierarchical partial-pooling model is preferred for confirmatory
pathogen-site comparison.
X. Final evidence hierarchy and next research steps
What the present evidence supports
The first negative is the main bottleneck
P → N1 is only 16.78%.
One negative is fragile
N1 → P occurs in 44.00%.
Two negatives are strong but incomplete
N2 → C is 73.10%, while N2 → P remains 26.90%.
Two hidden regimes improve the model
AICc, BIC, and patient-level prediction improve over the MM.
The biological gradient is coherent
Posterior colonization declines from P through C.
The simple two-state family is inadequate
Pooling, memorylessness, and predictive diagnostics fail.
What the present evidence does not establish
Unsupported statement
Why it remains unsupported
“16.78% is a weekly clearance probability.”
It is calculated per next included culture, not automatically per week.
“42.03% of patients recurred.”
42.03% is a transition-level C → P result.
“C proves biological eradication.”
C is an operational state defined by three qualifying negatives.
“HMM sensitivity and specificity are validated test accuracy.”
They are internally estimated emission parameters without an external gold standard.
“R1 and R2 identify specific pathogens or patient phenotypes.”
No covariate model has yet explained regime membership.
“Two regimes are definitively the true number.”
The recommendation remains provisional until repeated patient-level CV is completed.
Recommended research sequence
Primary explanation
Four-State Operational MM
Predictive extension
Two-regime Four-State switching HMM with repeated patient-level cross-validation
Biological sensitivity analysis
Positive-conditioned biological HMM with explicit initial-state sensitivity
Heterogeneity analysis
Hierarchical pathogen-site partial-pooling model
Time-aware recurrence analysis
Semi-Markov, continuous-time, or recurrent-event model with censoring
Data audit closure
Resolve the 50-, 19-, and 30-observation differences and review eight preparation errors
nGeneMDRO 최종 분석: Markov model과 hidden Markov model의 시각적 해석
필수 분석 실행
완료
Data pipeline 3/3 Core analyses 6/6
주 모형
Four-State MM
P → N1 → N2 → C
예측 확장 모형
2-regime HMM
AICc와 BIC에서 우세
Biological HMM
Exploratory
Initial-state sensitivity가 높음
Data provenance
검토 필요
50 + 19 + 30 observation 차이
통합 결론: MDRO clearance는 단순한 positive-to-negative event가 아니라
negative evidence가 단계적으로 누적되는 과정입니다. 가장 큰 장벽은 첫 qualifying negative를
얻는 것입니다. 두 번의 qualifying negative가 누적되면 confirmed clearance로 진행할 가능성이
상당히 높아지지만, 여전히 의미 있는 positive-reset risk가 남습니다. 따라서 Four-State MM은
primary explanatory model로, 2-hidden-regime switching HMM은 predictive extension으로
사용하는 것이 가장 타당합니다.
I. 전체 연구 지도의 시각화
분석 구조를 한눈에 보기
Raw data
2,703
명시적으로 parsing된 observation
→
Operational reconstruction
P · N1 · N2 · C
관찰된 evidence-history state
→
Hidden transition regime
R1 · R2
Persistence-prone과 clearance-progressing
+
Biological HMM
Clear ↔ Colonized
Latent biological-state sensitivity analysis
→
Interpretation
설명 · 예측 · audit
Research use이며 임상 release validation이 아님
모형별 domain coverage map
Research domain
Four-State MM
Switching HMM
Biological HMM
Simple two-state family
세 번 연속 음성 operational rule
주 모형
그대로 보존
간접적
표현하지 못함
N1 또는 N2 후 positive reset
직접 추정
Regime별 추정
주된 분석 대상이 아님
이력이 소실됨
숨은 transition heterogeneity
모델링하지 않음
핵심 분석 대상
다른 종류의 hidden concept
충분히 모델링하지 못함
Latent biological colonization
주장하지 않음
주장하지 않음
핵심 분석 대상
Reference estimate만 제공
Protocol의 투명한 해석
가장 높음
중간
낮음
높으나 구조적으로 부적절
현재 역할
Primary explanatory model
Predictive extension
Exploratory sensitivity analysis
Reference 및 assumption-failure model
II. Data provenance와 cohort flow
Observation reconciliation은 하나의 손실이 아니라 단계별 chain
2,772
Manuscript stated total
−50 →
Arithmetic gap
2,722
Pathogen subtotal
−19 →
Parser gap
2,703
Parsed observations
−30 →
Reconciliation
2,673
Prepared observations
50개, 19개, 30개의 차이는 서로 다른 provenance 단계에서 발생합니다. 이를 하나의
설명되지 않은 99개 차이로 합치면 안 되며 각각 별도로 추적되어야 합니다.
Sequence cohort funnel
Cohort 단계
Sequence
환자
이전 단계 대비 유지
감소 이유
Prepared patient-pathogen-site sequences
321
98
—
모든 prepared sequence
Observed-positive Four-State cohort
234
91
72.90%
87개에서 observed 또는 documented positive evidence가 없음
Switching-HMM comparison cohort
198
79
84.62%
36개에서 operational observation이 3개 미만
III. Four-State Operational Markov model
Operational state machine
P
Positive status
진행 중인 negative run 없음
16.78%
→
첫 qualifying negative
N1
First negative
Qualifying negative 1회
55.64%
→
두 번째 qualifying negative
N2
Two negatives
한 번 더 필요함
73.10%
→
세 번째 qualifying negative
C
Confirmed clearance
Local operational endpoint
P → P: 83.22% Positive persistence
N1 → P: 44.00% 음성 1회 후 reset
N2 → P: 26.90% 음성 2회 후 reset
C → P: 42.03% Clearance 이후 positive transition
Negative evidence가 누적될수록 forward progression이 쉬워짐
현재 상태
Forward progression
Positive persistence 또는 reset
임상·연구적 해석
P
P → N1: 16.78%
P → P: 83.22%
첫 qualifying negative가 가장 큰 bottleneck입니다.
N1
N1 → N2: 55.64%
N1 → P: 44.00%
음성 한 번은 의미가 있으나 불안정합니다.
N2
N2 → C: 73.10%
N2 → P: 26.90%
음성 두 번은 강한 evidence이나 confirmation은 여전히 필요합니다.
각 상태에서 다음 100개 transition을 생각하면
P에서 출발
83개가 P 유지17개가 N1 도달
N1에서 출발
44개가 P로 reset56개가 N2 도달
N2에서 출발
27개가 P로 reset73개가 C 확인
중단 없는 성공과 eventual clearance는 서로 다른 질문
6.82% · 중단 없는 clean-run product
Persistence, reset, 다음 재시도 없이 P → N1 → N2 → C로 바로 진행하는 경로입니다.
37.61% · 관찰기간 중 결국 C에 도달
Eligible sequence 234개 중 88개가 persistence, reset, 반복 시도를 포함하여 C에 한 번 이상
도달했습니다.
Evidence ladder의 시간적 경과
Clearance onset은 사후적으로 정의됩니다. 첫 번째 음성이 성공한 run의 시작이었다는 사실은
이후 두 번째와 세 번째 qualifying negative가 관찰된 뒤에야 확인할 수 있습니다.
IV. Clearance 이후 recurrence
Recurrence denominator tree
88
Cleared sequence
C에 한 번 이상 도달
→
39
Followed sequence
C 이후 transition이 존재
→
21
Recurrent sequence
C → P event가 한 번 이상 존재
43
Cleared patient
하나 이상의 sequence가 C 도달
→
27
Followed patient
C 이후 observation 존재
→
17
Recurrent patient
하나 이상의 recurrent sequence
다섯 개의 percentage는 다섯 개의 다른 질문
Endpoint
분자 / 분모
결과
측정하는 질문
C → P transition
29 / 69
42.03%
C에서 출발한 transition 중 다음 상태가 P였던 비율
Followed cleared sequence
21 / 39
53.85%
C 이후 follow-up이 있었던 sequence 중 recurrence
All cleared sequence
21 / 88
23.86%
C에 도달한 모든 sequence 중 관찰된 recurrence
Followed cleared patient
17 / 27
62.96%
C 이후 follow-up이 있었던 환자 중 recurrence
All cleared patient
17 / 43
39.53%
하나 이상의 C sequence를 가진 모든 환자 중 관찰된 recurrence
하나의 percentage를 보편적인 “recurrence rate”로 부를 수 없습니다. 모든 recurrence 결과에는
분석 단위, follow-up 조건, 분자, 분모, 관찰기간이 함께 제시되어야 합니다.
첫 관찰 recurrence까지의 시간
Lower quartile
7일
Median
11일
Upper quartile
31일
이른 positive return은 residual colonization의 intermittent detection, sampling variability,
false-negative run 또는 실제 reacquisition을 모두 포함할 수 있습니다. 현재 모형은 이러한
mechanism을 구분하지 못합니다.
V. Switching-HMM order 선택
Zero에서 five hidden regime까지의 information criteria
2-regime이 권장되는 이유
AICc winner
2 regimes
2164.659
BIC winner
2 regimes
2250.204
Single-split CV winner
4 regimes
Log loss 0.482243
제외된 후보
5 regimes
수렴하지 않음
판단 기준
2 regimes
4 regimes
해석
AICc
2164.659
2193.655
2-regime이 우세합니다.
BIC
2250.204
2415.218
2-regime이 매우 강하게 우세합니다.
CV log loss
0.486243
0.482243
4-regime의 한 번의 split 개선은 약 0.82%입니다.
Minimum regime occupancy
31.64%
13.15%
2-regime의 각 상태가 더 충분한 observation을 포함합니다.
Minimum regime separation
34.13%
10.74%
2-regime transition profile의 구분이 더 명확합니다.
1-regime candidate는 structural sanity check
Four-State MM
=
HMM · 1 hidden regime
=
동일한 likelihood, AICc, BIC, CV, transition matrix
이러한 정확한 일치는 direct Four-State MM이 switching-HMM family 안에 올바르게
nested되어 있음을 확인합니다.
VI. 두 개의 hidden transition regime
Regime별 operational pathway
R1 · Persistence/reset-prone
P7.55% →N128.51% →N242.07% →C
N1 → P: 71.20%
N2 → P: 56.97%
C → P: 83.54%
Regime persistence R1 → R1: 97.24%
R2 · Clearance-progressing
P54.34% →N169.77% →N279.30% →C
N1 → P: 29.56%
N2 → P: 20.49%
C → C: 61.45%
Regime persistence R2 → R2: 99.67%
두 transition environment의 직접 비교
R1과 R2는 transition pattern을 설명하는 label입니다. 특정 pathogen, 특정 환자 phenotype,
중증도 group 또는 biological Clear/Colonized state로 증명된 것이 아닙니다.
VII. Biological Clear/Colonized HMM
Operational evidence와 latent colonization은 같은 방향으로 움직임
P
92.53%
≫
N1
25.89%
>
N2
3.01%
>
C
0.94%
Gradient는 일관되지만 절대 확률은 model-dependent
비교적 안정적인 결론
두 biological HMM 모두 P에서 N1, N2, C로 갈수록 posterior colonization이 급격히
감소합니다.
아직 exploratory한 부분
정확한 posterior percentage, α, β, sensitivity, specificity는 hidden-state initialization과
model assumption에 상당한 영향을 받습니다.
Initial-state sensitivity
Initial-state profile
BIC
Stationary profile과의 차이
해석
Positive-conditioned baseline
2203.841
+26.133
Protocol-aligned primary profile이나 상대적 fit은 약함
Estimated initial state
2185.421
+7.713
Baseline distribution을 추정할 때 fit이 개선됨
Stationary initial state
2177.708
0
검토한 profile 중 가장 낮은 BIC
Emission parameter는 검사실 validation이 아님
HMM이 추정하는 것
각 latent state에서 positive 또는 negative result가 관찰될 확률
Longitudinal sequence를 가장 잘 설명하는 transition behavior
≠
아직 확립되지 않은 것
외부 gold standard로 검증된 culture sensitivity 또는 specificity
Predictive check는 MM 4개 중 2개, HMM 4개 중 4개에서 문제가 있으며 homogeneity와
memorylessness 가정도 강하게 기각됩니다.
두 모형 모두 primary model로는 부적절합니다.
Two-state model에서 사라지는 history
Simple two-state representation
Positive ↔ Negative
첫 번째 음성과 두 번째 연속 음성이 같은 state로 처리됩니다.
→
Four-State representation
P → N1 → N2 → C
임상적으로 정의된 evidence history가 직접 보존됩니다.
Diagnostic
결과
의미
Pathogen-site homogeneity
P = 3.547 × 10−21
하나의 pooled transition process가 지지되지 않습니다.
First-order memorylessness
P = 5.306 × 10−11
현재 positive 또는 negative만으로는 충분한 history를 표현하지 못합니다.
IX. Pathogen-site heterogeneity
2차원 progression map
가로축은 P에서 벗어나 첫 qualifying negative에 도달하는 능력을 나타냅니다. 세로축은 N2 이후
C를 확인할 확률을 나타냅니다. Bubble 크기는 sequence 수를 반영합니다.
Progression heat map
Pathogen-site
Sequence
P → N1
N1 → N2
N2 → C
시각적 pattern
CRE · Stool
67
9.50%
48.53%
59.38%
P에서 강한 persistence
CRE · Sputum
31
15.71%
51.61%
62.50%
느린 progression
CRE · Urine
35
18.75%
57.69%
74.07%
CRE stool보다 양호함
VRE · Stool
36
15.56%
46.67%
65.00%
N1 이후 reset이 많음
VRE · Urine
23
41.10%
75.00%
95.00%
강한 forward progression
MRAB · Sputum
14
67.86%
63.16%
90.91%
빠른 progression 신호이나 표본이 작음
MRPA · Sputum
9
66.67%
66.67%
60.00%
진입은 빠르지만 confirmation이 불확실함
MRPA · Urine
10
32.50%
76.92%
80.00%
양호한 progression이나 sparse data
Small stratum에서 나타나는 0% 또는 100%를 안정적인 population probability로 해석하면
안 됩니다. Confirmatory pathogen-site comparison에는 hierarchical partial-pooling model이
더 적절합니다.
X. 최종 evidence hierarchy와 다음 연구 단계
현재 evidence가 지지하는 결론
첫 음성이 가장 큰 bottleneck
P → N1은 16.78%에 불과합니다.
음성 한 번은 불안정함
N1 → P가 44.00%입니다.
음성 두 번은 강하지만 불완전함
N2 → C는 73.10%이나 N2 → P도 26.90%입니다.
2-hidden regime이 모형을 개선함
AICc, BIC, patient-level prediction이 MM보다 개선됩니다.
Biological gradient가 일관됨
Posterior colonization이 P에서 C로 갈수록 감소합니다.
Simple two-state family가 부적절함
Pooling, memorylessness, predictive diagnostic이 모두 실패합니다.
현재 evidence가 확립하지 못하는 주장
지지되지 않는 주장
아직 지지되지 않는 이유
“16.78%는 주간 clearance probability이다.”
다음 included culture 1회당 계산된 값이며 자동으로 주간 확률을 뜻하지 않습니다.
“42.03%의 환자가 recurrence하였다.”
42.03%는 transition-level C → P 결과입니다.
“C는 biological eradication을 증명한다.”
C는 qualifying negative 3회로 정의된 operational state입니다.
“HMM sensitivity와 specificity는 검증된 검사 정확도이다.”
External gold standard 없이 내부적으로 추정된 emission parameter입니다.
“R1과 R2는 특정 pathogen 또는 환자 phenotype을 식별한다.”
아직 regime membership을 설명하는 covariate model이 없습니다.
“2-regime이 definitively true number이다.”
Repeated patient-level CV가 완료될 때까지 권고는 provisional합니다.
권장 연구 순서
Primary explanation
Four-State Operational MM
Predictive extension
Repeated patient-level cross-validation을 포함한 2-regime Four-State switching HMM
Heterogeneity analysis
Hierarchical pathogen-site partial-pooling model
Time-aware recurrence analysis
Censoring을 반영한 semi-Markov, continuous-time 또는 recurrent-event model
Data audit closure
50개, 19개, 30개 observation 차이를 해결하고 preparation error 8개를 검토
Written on July 28, 2026
Interpreting the nGeneMDRO complete analysis results: a visual guide (Written July 30, 2026)
Scope.
This interpretation is based on the complete analysis report generated from
RawDataset.zip and the linked patient-outcome dataset. The report covers
98 patients, 2,703 parsed culture observations, 2,673 reconciled observations,
321 prepared patient–pathogen–site sequences, and 29 analysis sections.
I. Executive interpretation
One-page visual reading map
98
Patients
2,703
Parsed cultures
234
Four-state sequences
16.78%
P → N1 bottleneck
73.10%
N2 → C confirmation
2
Preferred hidden regimes
Raw cultures
2,703 observations
→
Operational states
P → N1 → N2 → C
→
Durability
C → P recurrence
→
Patient endpoint
Death or censoring
→
Model layer
Multi-state and HMM
The primary interpretable result is the protocol-aligned four-state model.
The first qualifying negative is the principal bottleneck. Additional
qualifying negatives progressively strengthen the evidence for confirmed
local clearance. Recurrence remains frequent when surveillance continues,
death-aligned cultures do not show the strong terminal rise in negativity
seen before non-death censoring, and two hidden transition regimes improve
prediction without replacing the observed operational states.
Evidence domain map
The analyses differ along two dimensions: whether the quantity is directly
reconstructed from observed cultures or generated by a statistical model,
and whether its present evidence is relatively supported or exploratory.
The pathway widens after each qualifying negative.
Protocol-aligned evidence
How many sequences eventually reached C?
88 of 234, or 37.61%
Repeated attempts produce more eventual clearances than one clean run.
Cumulative descriptive endpoint
Was confirmed C always durable?
21 of 39 followed cleared sequences recurred
Durability is limited when surveillance continues.
Follow-up-dependent description
Did negativity rise near death?
Paired change −8.53 percentage points
No death-terminal increase comparable with censoring was seen.
Observational, not causal
What occurred before non-death censoring?
Paired change +18.29 percentage points
Negativity rose markedly near discharge, transfer, withdrawal, or administrative censoring.
Consistent but heterogeneous comparator
What was the 90-day outlook from P?
C 16.29%; Death 20.21%; unresolved 63.50%
Most episodes remained in P, N1, or N2 at 90 days.
Exploratory model-based probability
Which hidden-regime order was preferred?
Two hidden regimes
Two regimes gave the best balance of fit, prediction, occupancy, and parsimony.
Secondary predictive layer
What the report establishes and what it does not establish
Reasonably supported
The first qualifying negative is the main operational bottleneck.
N1 and N2 retain clinically relevant evidence history.
Death-aligned and censoring-aligned terminal trajectories differ.
The two-regime switching HMM predicts operational transitions better than the single-regime baseline.
Not established
C is not proven biological eradication and is not automatically PLC or PtLC.
Terminal illness is not proven to cause the culture trajectory.
No single recurrence percentage represents patient incidence.
Hidden regimes are not validated biological phenotypes or patient subtypes.
The model outputs are not validated isolation-release or treatment thresholds.
II. Data foundation and cohort flow
Dataset composition
98
Source files
2,703
Explicit observations
2,673
Prepared observations
321
Prepared sequences
CRE contributes most observations and therefore strongly influences pooled results.
Stool and urine dominate the specimen distribution; blood contains only seven observations.
CRE accounts for 1,771 of 2,703 observations, while stool accounts for
1,382 observations. The pooled analysis therefore primarily reflects CRE
and stool or urine surveillance. Blood and several wound strata remain too
sparse for stable standalone inference.
Observation reconciliation waterfall
2,772
Manuscript-stated episodes
→
−50
2,722
Manuscript pathogen subtotal
→
−19
2,703
Explicit parser count
→
−30
2,673
Prepared observations
Difference
Size
Where it arises
Required interpretation
Manuscript arithmetic gap
50
Inside the manuscript
Source inconsistency; it must remain separate.
Manuscript-to-parser gap
19
Source reconstruction
Requires source-line attribution.
Parser-to-preparation reduction
30
Same-day reconciliation and sequence preparation
Requires a deterministic observation-level ledger.
These are three distinct provenance problems. Combining them into one
discrepancy would hide whether the correction belongs to the manuscript,
source-file parser, or sequence-preparation rules.
The four-state analysis represents 72.90% of prepared sequences. The
switching-HMM comparison represents 61.68%. Model-order findings therefore
apply to a more intensively observed subset rather than the complete
prepared cohort.
Pathogen–site domain coverage map
Bubble size represents the number of eligible sequences. Bubble color represents
the fraction of sequences that reached C. Zero-sequence cells are omitted.
The map distinguishes broad, data-rich domains from visually impressive
but sparse cells. For example, CRE stool contains 67 sequences and only
16 reached C, whereas several 100% transition values arise from one or two
sequences and should not be read as stable superiority.
Data-audit implications
Parser
5 informational issues, 0 warnings, 0 errors.
Sequence preparation
22 issues, including 8 errors.
Outcome import
126 parser issues without a displayed severity ledger.
Completion of every model does not resolve data provenance. Final scientific
interpretation should remain provisional until the 30 removed or merged
observations, all 8 sequence-preparation errors, and the 126 outcome-parser
issues can be traced to explicit rules or source lines.
III. Four-state operational clearance
Operational state machine
P
Positive
First qualifying negative
16.78%
→
N1
One negative
Second qualifying negative
55.64%
→
N2
Two negatives
Third qualifying negative
73.10%
→
C
Confirmed local clearance
N1 → P: 44.00%
A positive culture resets one negative.
N2 → P: 26.90%
Two negatives remain incomplete evidence.
C → P: 42.03%
Later positivity can occur after confirmed local clearance.
The minimum interval between qualifying negatives is three days. A positive
result before C resets the sequence to P. Clearance onset is assigned to the
first negative in the final successful run, while confirmation occurs at the
third qualifying negative.
Bottleneck profile
The forward probability increases sharply after negative evidence begins to accumulate.
Source state
Forward transition
Persistence or reset
Main interpretation
P
P → N1: 299/1,782 = 16.78%
P → P: 83.22%
Entering the negative sequence is difficult.
N1
N1 → N2: 153/275 = 55.64%
N1 → P: 44.00%
One negative is fragile.
N2
N2 → C: 106/145 = 73.10%
N2 → P: 26.90%
Two negatives are strong but not definitive.
C
C → C: 40/69 = 57.97%
C → P: 42.03%
Confirmed local clearance is not always durable.
Clean-run probability versus eventual clearance
6.82%
Uninterrupted clean run
P → N1 → N2 → C without persistence, reset, or another attempt.
37.61%
Eventually reached C
88 of 234 sequences reached C after all observed attempts.
\(0.1678 \times 0.5564 \times 0.7310 = 0.0682\)
The 6.82% value is a single-path probability. The 37.61% value is a
cumulative observed endpoint fraction. Repeated attempts explain why the
latter is much larger.
Timing ribbon and stage occupancy
20.0 days
Median time to first N1
→
38.0 days
Median time to N2
→
48.5 days
Median confirmation time
P
1,906 observations
N1
300
N2
153
C
146
The median clearance-onset time of 34.5 days may appear earlier than the
median N2 time of 38.0 days because the two summaries can involve different
eligible subsets and unsuccessful earlier attempts. Each timing denominator
should therefore remain explicit.
Stratum-level interpretation
Relatively favorable observed pathway
VRE urine: P → N1 41.10%; N1 → N2 75.00%; N2 → C 95.00%.
23 sequences; still requires uncertainty-aware interpretation.
Marked initial bottleneck
CRE stool: P → N1 9.50%; N1 → N2 48.53%; N2 → C 59.38%.
67 sequences; a large and influential stratum.
Sparse apparent perfection
Several wound cells show 100% transitions based on one or two events.
These should remain descriptive rather than inferential.
IV. Recurrence after confirmed clearance
Recurrence denominator tree
88 cleared sequences
↓
39 followed sequences
At least one counted post-clearance transition
↓
21 recurrent sequences
53.85% of followed sequences
↓
49 sequences without counted follow-up
55.68% of all cleared sequences
↓
Recurrence status incompletely observable
Lack of observation is not proof of durability
Why there is no single recurrence rate
The estimate changes because the unit and follow-up requirement change.
Endpoint
Numerator / denominator
Estimate
What it measures
Transition-level C → P
29 / 69
42.03%
Positive next transition among all transitions observed from C.
Followed sequence recurrence
21 / 39
53.85%
At least one recurrence among followed cleared sequences.
All-cleared sequence recurrence
21 / 88
23.86%
Observed recurrence among all sequences reaching C.
Followed patient recurrence
17 / 27
62.96%
At least one recurrent sequence among followed cleared patients.
All-cleared patient recurrence
17 / 43
39.53%
Observed recurrence among all patients with at least one C sequence.
Patient-level follow-up funnel
43 cleared patients
→
27 followed patients
→
17 recurrent patients
16 of 43 cleared patients had no counted post-clearance
transition in any cleared sequence.
The followed denominator may be enriched for patients who remained under
surveillance. The all-cleared denominator, in contrast, treats absence of
documented recurrence as no observed event. Neither is a surveillance-independent
patient incidence estimate.
Recurrence timing landscape
Most first recurrences occurred early, but a long tail extended to 301 days.
11 days
Median time to first recurrence
7–31 days
Interquartile range
6 sequences
More than one recurrence
Appropriate recurrence conclusion
Confirmed local clearance was not always durable under continued surveillance.
The present report does not estimate a surveillance-independent patient
recurrence incidence. A recurrent-event model with censoring, time at risk,
and patient clustering is required for that purpose.
V. Endpoint-aligned terminal analysis
Endpoint cohort flow
98 of 98 patients linked to an outcome record
No endpoint-date conflicts
↓
44 deaths
↓
34 patients in 12-week terminal models
↓
28 paired patients
Represented in both windows
↓
54 censoring endpoints
↓
43 patients in 12-week terminal models
↓
28 paired patients
Represented in both windows
The censoring group combines administrative censoring, discharge, transfer,
and withdrawal. It should not be interpreted as a biologically uniform
survivor-control group.
Weekly reverse-time trajectories
The final four weeks are visually shaded. Censoring-aligned negativity rises,
while the death-aligned trajectory remains low.
Weekly values describe cultures that were actually collected. An uncollected
or unobtainable specimen is not converted into a negative result.
Paired terminal-window contrast
Death-aligned
Earlier window → terminal window
↓
−8.53 percentage points
95% interval: −15.00 to −2.74 percentage points
28 paired patients
Censoring-aligned
Earlier window → terminal window
↑
+18.29 percentage points
95% interval: +6.99 to +28.12 percentage points
28 paired patients
The central terminal finding is a contrast between endpoints. The death-aligned
course lacks the strong rise in negativity observed before non-death censoring.
The selected boundary is a two-segment description of timing, not evidence
that terminal illness caused a biological switch.
Three adjusted model lenses
Mixed-effects logistic
Linear four-week trend with patient random intercept.
Death trend OR: 0.896
Death-versus-censoring ratio: 0.455
Patient CV log loss: 0.654
Generalized additive mixed model
Nonlinear curve with patient random intercept.
Patient CV log loss: 0.629
Improvement: 0.0251
Preferred frequentist prediction model
Bayesian hierarchical model
Probability statements under a Laplace approximation.
Death change: −2.43 percentage points
P(death increase): 20.82%
P(death change > censoring change): 0.00%
Adjusted result
Estimate
Interpretation
Four weeks closer to censoring
OR 1.968, 95% interval 1.601–2.420
Negative-culture odds rise strongly.
Four weeks closer to death
OR 0.896, 95% interval 0.689–1.164
No convincing rise; the direction is slightly downward.
Death-versus-censoring trend ratio
OR 0.455, 95% interval 0.325–0.636
The endpoint trajectories differ substantially.
Sampling intensity
OR 1.485, 95% interval 1.034–2.135
Observation frequency may be informative rather than neutral.
Observation-process limitation
Clinical status
→
Whether a specimen is obtained
→
Which site is sampled
→
Observed positive or negative result
Treating missing specimens as neither positive nor negative is correct, but
it does not eliminate informative missingness. Future analyses should model
the probability of specimen collection, separate censoring reasons, and
stratify the terminal curves by specimen site.
VI. Competing-risk multi-state analysis
First-event model structure
P
⇄
N1
⇄
N2
Confirmed local C
Absorbing first event
Death
Competing absorbing event
The model follows one patient–pathogen–site episode through P, N1, and N2
until confirmed local C or Death occurs first. Post-clearance recurrence is
deliberately outside this first-event model.
Episode outcome accounting
88 C first
37.61%
83 Death first
35.47%
63 censored
26.92%
234
Episodes
91
Unique patients
400 / 400
Valid patient-cluster bootstraps
Episode counts are not interchangeable with patient counts. A single death
can terminate several active pathogen–site episodes contributed by the same
patient.
Transition-intensity map
Current state
Forward rate per 100 exposure-days
Reset rate
Death rate
Visual reading
P
P → N1: 1.0336
Remaining P is continued exposure
P → Death: 0.2826
The exit from persistent positivity is slow.
N1
N1 → N2: 3.4464
N1 → P: 2.8765
N1 → Death: 0.2442
Progression and reset remain in close competition.
N2
N2 → C: 3.7084
N2 → P: 1.3064
N2 → Death: 0.1686
Confirmed C dominates the next modeled exit.
P → N1
1.0336 per 100 exposure-days
N1 → N2
3.4464 per 100 exposure-days
N2 → C
3.7084 per 100 exposure-days
These values are exposure-time rates, not probabilities per culture. A
three-day interval and a fourteen-day interval contribute different time
at risk.
Eventual C-before-Death probability
Accumulated negative evidence shifts the eventual first-event balance toward C.
53.40%
Starting from P
68.00%
Starting from N1
85.00%
Starting from N2
Time-dependent horizon map
Fixed-horizon probabilities preserve the unresolved P/N1/N2 fraction.
Starting from P
Confirmed C
Death
Still P/N1/N2
30 days
2.41%
7.91%
89.68%
90 days
16.29%
20.21%
63.50%
180 days
32.47%
31.78%
35.75%
365 days
47.01%
42.08%
10.91%
“C before Death is 53.40% from P” and “C is 16.29% at 90 days
from P” answer different questions. The first is an eventual absorption
probability; the second preserves episodes that remain unresolved at a
fixed time.
Model boundary
What the model preserves
Irregular exposure time
Competing first events
Patient-cluster uncertainty
What the model does not preserve
Exact interval-censored transition times
Duration-dependent transition intensities
Post-C recurrent events
Time-varying surveillance intensity
VII. HMM findings and model selection
Biological HMM stage gradient
Every profile shows a monotonic decline, but the magnitude changes with the initial-state assumption.
The positive-conditioned primary profile gives a mean latent-colonization
probability of 92.53% in P, 25.50% in N1, 2.96% in N2, and 0.94% in C.
The direction is coherent, but this gradient is generated from the same
culture history used to define the operational stages and is not an
independent validation of eradication.
Initial-state sensitivity
Initial-state profile
P
N1
N2
C
BIC
Positive-conditioned baseline
92.53%
25.50%
2.96%
0.94%
2,206.435
Estimated initial state
86.54%
24.47%
3.38%
0.63%
2,187.042
Stationary initial state
86.56%
24.67%
3.45%
0.63%
2,179.351
Design-aligned primary
→
Alternative BIC improves by 27.084
→
Biological interpretation remains exploratory
Core parameters and stage posteriors change materially, and two distinct
likelihood modes were detected. The report therefore appropriately labels
the evidence as Exploratory and the initial-state sensitivity
as Highly Sensitive.
Switching-HMM order comparison
The two-regime model minimizes BIC, while four regimes achieve only a small additional CV improvement.
Model
Converged
AICc
BIC
CV log loss
Minimum occupancy
Minimum separation
Four-state MM / one regime
Yes
2,254.700
2,288.967
0.506774
100.00%
NE
Two regimes
Yes
2,164.659
2,250.204
0.486243
31.64%
34.13%
Three regimes
Yes
2,173.920
2,321.938
0.483468
27.59%
12.57%
Four regimes
Yes
2,193.655
2,415.218
0.482243
13.15%
10.74%
Five regimes
No
2,224.257
2,530.284
0.483921
5.15%
12.69%
Two-regime transition-domain map
Regime 2 has a broadly larger progression domain, while Regime 1 is dominated by return toward P.
Regime 1 · lower progression
P → N1: 7.55%
N1 → N2: 28.51%
N2 → C: 42.07%
C → C: 16.46%
Observed occupancy: 68.36%
Regime 2 · higher progression
P → N1: 54.34%
N1 → N2: 69.77%
N2 → C: 79.30%
C → C: 61.45%
Observed occupancy: 31.64%
These regimes describe transition patterns. They should not be named
“persistent patients” and “clearing patients.” The occupancy and switching
asymmetry may reflect sequence phase or time evolution rather than fixed
biological patient classes.
Why two regimes were selected
Best AICc
+
Best BIC
+
4.05% CV improvement over MM
+
Acceptable occupancy and separation
→
Preferred: two regimes
Four regimes improve CV log loss over two regimes by only 0.0040, approximately
0.82%, while worsening BIC by about 165 points and reducing regime separation.
Repeated patient-level cross-validation remains necessary to assess order stability.
Why the simple two-state family remains a reference
Transition homogeneity
p = 2.536 × 10−21
Memorylessness
p = 3.657 × 10−11
Markov checks
2 of 4 flagged
HMM checks
4 of 4 flagged
The two-state HMM improves AICc, BIC, and cross-validated log loss relative
to the two-state observed Markov model. Relative improvement, however, does
not establish absolute adequacy. The report’s verdict, “Neither model adequate,”
is therefore appropriate.
VIII. Reliability review and output defects
Issue-priority map
High impact · lower urgency
Repeated cross-validation and external HMM validation
High impact · moderate urgency
Separate censoring mechanisms and model specimen collection
High impact · immediate
Data reconciliation, 8 sequence errors, 126 outcome-parser issues
Moderate impact · lower urgency
Additional report navigation and collapsible appendices
Moderate impact · moderate urgency
Differentiate the two biological HMM summaries
Moderate impact · immediate
Repair 0–100% prediction intervals and sparse coefficients
Lower impact · lower urgency
Terminology and visual-label refinement
Lower impact · moderate urgency
Display all timing denominators explicitly
Lower impact · immediate
Escape vertical bars in the recurrence-event table
Priority corrections
Priority
Issue
Why it matters
Recommended correction
Critical
Observation gaps of 50, 19, and 30 remain unresolved.
Source completeness cannot be inferred from successful execution.
Create an observation-level reconciliation ledger.
Critical
Eight sequence-preparation errors remain.
All operational and downstream states depend on sequence construction.
List the affected sequence, source line, rule, and inclusion decision.
Critical
Outcome import contains 126 parser issues.
Terminal and competing-risk models depend on endpoint integrity.
Expose issue severity and a patient-level audit table.
High
Mixed-model weekly intervals are 0% to 100%.
The intervals are not informative and may reflect a variance-calculation problem.
Separate population confidence, new-patient prediction, and conditional intervals.
High
Blood-versus-stool OR is approximately 52.6 million.
Seven blood observations create a sparse-data or separation problem.
Suppress as NE, combine levels, or use an explicitly penalized model.
High
Twenty-one linked patients are absent from terminal models without a flow table.
The reason for exclusion cannot be identified.
Add one explicit exclusion reason per patient.
Moderate
Sparse cells display 100% values.
Visual prominence may be mistaken for certainty.
Display numerator, denominator, and estimability warnings.
Recurrence-event table formatting defect
Current output
1022|CRE|Stool
Markdown interprets the vertical bars as table-column separators.
Safe output
1022\|CRE\|Stool
1022 · CRE · Stool
Either escape the delimiter or replace it with a non-table character.
Report-architecture improvement
Current reading burden
Long uninterrupted technical sections
Critical and exploratory results mixed together
Very long episode and sequence tables in the main flow
Percentages without immediate denominator context
→
Recommended visual architecture
One-page evidence dashboard first
One visual question per section
Numerator, denominator, unit, and follow-up beside each percentage
Long audits moved to collapsible appendices
Automatic warnings for sparse or unstable estimates
IX. Research conclusions and next priorities
Major strengths
Protocol fidelity
The four-state model directly encodes the three-negative rule.
Denominator transparency
Transition, sequence, and patient recurrence are kept separate.
Complete outcome linkage
All 98 culture patients have verified endpoints.
Cluster-aware uncertainty
Repeated observations are addressed at the patient level.
Model guardrails
Operational, biological, causal, and clinical meanings are separated.
Multiple complementary views
Descriptive, predictive, Bayesian, and competing-risk questions remain distinct.
Supported substantive conclusions
First negative
Main bottleneck
→
N1 and N2
Meaningful evidence history
→
Third negative
Meaningful confirmation
→
After C
Durability remains uncertain
The death-aligned trajectory differs substantially from the non-death
censoring trajectory, but neither the reverse-time models nor the adjusted
models establish a causal terminal-illness mechanism.
Recommended order of further work
1. Close the data audit
Resolve every source, parser, preparation, and outcome issue.
2. Repair uncertainty displays
Correct 0–100% intervals and suppress unstable sparse coefficients.
3. Stabilize model selection
Repeat patient-level CV and bootstrap regime-order selection.
4. Separate censoring mechanisms
Analyze discharge, transfer, withdrawal, and administrative endpoints separately.
5. Model specimen collection
Evaluate informative sampling and site-specific missingness.
6. Develop recurrent-event models
Estimate first and repeated recurrence with time at risk.
7. Extend the multi-state model
Test semi-Markov and interval-censored formulations.
8. Seek external validation
Validate biological HMM emissions, regimes, and outcome probabilities elsewhere.
Manuscript-ready substantive conclusion
In this surveillance cohort, the principal operational barrier to confirmed
local clearance was obtaining the first qualifying negative culture. Once
negative evidence accumulated, progression became increasingly favorable,
supporting retention of N1 and N2 as distinct evidence-history states and
preserving the third negative as a meaningful confirmation step. Confirmed
clearance was not uniformly durable under continued surveillance, although
recurrence estimates depended strongly on the unit of analysis and the
availability of post-clearance follow-up. Death-aligned cultures showed no
terminal rise in negativity comparable with the pronounced increase observed
before non-death censoring, but the observational design and changing specimen
availability preclude a causal interpretation. The four-state operational
model should remain the primary interpretable analysis; the two-regime
switching HMM may be retained as a secondary predictive layer; biological
HMM parameters and competing-risk probabilities should remain explicitly
model-based and exploratory.
nGeneMDRO 전체 분석 결과의 시각적 해석
분석 범위.
본 해석은 RawDataset.zip과 연계된 환자 종점 자료에서 생성된
전체 분석 보고서를 기반으로 한다. 보고서에는 환자 98명, 파싱된 배양
관찰 2,703건, 조정 후 관찰 2,673건, 환자–병원체–검체 부위 단위의
준비된 시퀀스 321개, 총 29개 분석 섹션이 포함되어 있다.
I. 핵심 해석
한 페이지 시각적 독해 지도
98
환자
2,703
파싱 배양
234
4상태 시퀀스
16.78%
P → N1 병목
73.10%
N2 → C 확인
2
선호 숨은 체제
원자료 배양
관찰 2,703건
→
운영 상태
P → N1 → N2 → C
→
지속성
C → P 재양성
→
환자 종점
사망 또는 검열
→
모델 층
다상태 모델과 HMM
가장 해석 가능한 1차 결과는 프로토콜에 직접 대응하는 4상태 모델이다.
첫 번째 적격 음성을 얻는 단계가 가장 큰 병목이며, 적격 음성이
추가될수록 확인된 국소 음전의 근거가 강해진다. 추적이 지속된
경우 재양성은 흔하였고, 사망 전 배양에서는 비사망 검열 전과
같은 강한 음성률 상승이 나타나지 않았다. 2개의 숨은 전이 체제는
예측을 개선하지만 관찰된 운영 상태를 대체하지 않는다.
근거 영역 지도
각 분석은 관찰 자료에서 직접 재구성되는 정도와 통계적 모델에
의존하는 정도가 다르며, 현재 근거 역시 비교적 지지되는 영역부터
탐색적 영역까지 구분된다.
비교적 지지됨 · 직접 재구성
종점 연계4상태 전이재양성 분모코호트 흐름
비교적 지지됨 · 모델 기반
GAMM 예측2체제 전환 HMM군집 부트스트랩
주의 필요 · 직접 관찰
주별 말기 음성률희소 층음전 후 추적
탐색적 · 모델 기반
생물학적 HMM 모수C-선행-사망 흡수확률사망 변화점2상태 생물학적 해석
핵심 결과 한눈에 보기
질문
주요 결과
시각적 의미
근거 역할
음전 경로에서 가장 어려운 단계는 어디인가?
P → N1: 16.78%
첫 음성으로 들어가는 통로가 가장 좁다.
1차 기술 결과
음성 근거가 누적될수록 의미가 커지는가?
N1 → N2: 55.64%; N2 → C: 73.10%
적격 음성이 추가될수록 다음 단계 통로가 넓어진다.
프로토콜 대응 근거
최종적으로 C에 도달한 비율은 얼마인가?
234개 중 88개, 37.61%
재시도가 가능하므로 단일 성공 경로보다 최종 도달이 많다.
누적 기술 종점
확인 C는 항상 유지되었는가?
추적 시퀀스 39개 중 21개 재양성
추적이 계속될 때 지속성은 제한적이었다.
추적 의존 기술 결과
사망에 가까워질수록 음성률이 상승했는가?
짝지은 변화 −8.53%p
검열 전과 같은 말기 음성률 상승은 나타나지 않았다.
관찰적이며 비인과적
비사망 검열 전에는 어떤 변화가 있었는가?
짝지은 변화 +18.29%p
퇴원, 전원, 철회 또는 행정적 종점 전 음성률이 상승하였다.
일관되지만 비교군이 이질적
P에서 시작한 90일 전망은 어떠한가?
C 16.29%; 사망 20.21%; 미해결 63.50%
대부분은 90일에도 P, N1 또는 N2에 남아 있었다.
탐색적 모델 확률
숨은 체제 수는 몇 개가 적절했는가?
2개 숨은 체제
적합도, 예측력, 점유율, 단순성의 균형이 가장 좋았다.
2차 예측 층
입증되는 내용과 입증되지 않는 내용
비교적 지지되는 내용
첫 번째 적격 음성이 가장 큰 운영적 병목이다.
N1과 N2는 의미 있는 근거 이력을 담고 있다.
사망 정렬과 검열 정렬의 말기 경향은 다르다.
2체제 전환 HMM은 단일 체제 기준선보다 운영 상태를 잘 예측한다.
입증되지 않는 내용
C는 생물학적 완전 제거로 입증된 상태가 아니다.
말기 질환이 배양 경향을 유발했다는 인과관계는 입증되지 않았다.
하나의 재양성 백분율이 환자 발생률 전체를 대표하지 않는다.
숨은 체제는 검증된 환자 아형이나 생물학적 표현형이 아니다.
본 결과는 격리 해제 또는 치료 결정을 위한 검증 기준이 아니다.
II. 자료 기반과 코호트 흐름
자료 구성
98
원본 파일
2,703
명시적 관찰
2,673
준비된 관찰
321
준비 시퀀스
CRE가 전체 관찰의 대부분을 차지하므로 통합 결과에 큰 영향을 준다.
대변과 소변이 검체 구성을 지배하며 혈액 관찰은 7건뿐이다.
CRE는 2,703건 중 1,771건이고 대변 검체는 1,382건이다. 따라서
통합 결과는 주로 CRE와 대변 또는 소변 감시를 반영한다. 혈액 및
일부 상처 층은 독립적인 안정 추론을 하기에는 지나치게 희소하다.
관찰 수 조정 폭포도
2,772
원고 기재 전체 배양
→
−50
2,722
원고 병원체별 소계
→
−19
2,703
명시적 파서 집계
→
−30
2,673
준비된 관찰
차이
크기
발생 단계
필요한 해석
원고 산술 차이
50
원고 내부
원자료 불일치로 별도 유지해야 한다.
원고–파서 차이
19
원본 재구성
원본 문장 단위 추적이 필요하다.
파서–준비 감소
30
동일 날짜 조정과 시퀀스 준비
관찰 단위 결정 규칙 원장이 필요하다.
세 차이는 서로 다른 출처 문제이다. 하나의 총차이로 합치면 원고,
원본 파서, 시퀀스 준비 중 어느 단계에서 수정이 필요한지 알 수 없게 된다.
시퀀스 선택 퍼널
준비된 환자–병원체–부위 시퀀스 321개
환자 98명
↓
양성 관찰 기반 4상태 시퀀스 234개
환자 91명 · 시퀀스 87개 제외
↓
숨은 체제 비교 시퀀스 198개
환자 79명 · 추가 36개 제외
4상태 분석은 준비 시퀀스의 72.90%를 포함하고, 전환 HMM 비교는
61.68%를 포함한다. 따라서 모델 차수 결과는 전체 준비 코호트보다
관찰이 더 충분한 하위집단에 적용된다.
병원체–검체 부위 영역 지도
버블 크기는 적격 시퀀스 수, 색상은 C에 도달한 시퀀스 비율을 나타낸다.
시퀀스가 없는 셀은 표시하지 않았다.
이 지도는 자료가 충분한 넓은 영역과, 수치는 좋아 보이지만 표본이
지나치게 작은 영역을 구분한다. 예를 들어 CRE 대변은 67개 시퀀스를
포함하지만 16개만 C에 도달하였다. 반면 일부 100% 값은 한두 개의
시퀀스에 근거하므로 안정적인 우수성으로 해석해서는 안 된다.
자료 감사의 의미
파서
정보성 문제 5건, 경고 0건, 오류 0건.
시퀀스 준비
문제 22건, 그중 오류 8건.
종점 가져오기
심각도 원장 없이 파서 문제 126건.
모든 모델이 실행되었다는 사실은 자료 출처 문제가 해결되었다는
뜻이 아니다. 제거 또는 병합된 30건, 시퀀스 오류 8건, 종점 파서
문제 126건 각각이 명시적 규칙 또는 원본 문장에 연결되기 전까지
최종 과학적 해석은 잠정적으로 유지하는 편이 타당하다.
III. 4상태 운영적 음전
운영 상태 기계
P
양성
첫 적격 음성
16.78%
→
N1
음성 1회
두 번째 적격 음성
55.64%
→
N2
음성 2회
세 번째 적격 음성
73.10%
→
C
확인된 국소 음전
N1 → P: 44.00%
양성이 나오면 음성 1회가 초기화된다.
N2 → P: 26.90%
음성 2회도 완전한 확인은 아니다.
C → P: 42.03%
확인된 국소 음전 이후에도 다시 양성이 될 수 있다.
적격 음성 사이의 최소 간격은 3일이다. C에 도달하기 전 양성이
나오면 P로 초기화된다. 음전 시작은 최종 성공 구간의 첫 음성일이며,
확인은 세 번째 적격 음성일이다.
병목 구조
음성 근거가 누적될수록 전진 확률이 뚜렷하게 증가한다.
출발 상태
전진 전이
지속 또는 초기화
주요 의미
P
P → N1: 299/1,782 = 16.78%
P → P: 83.22%
음성 연속 과정에 진입하기 어렵다.
N1
N1 → N2: 153/275 = 55.64%
N1 → P: 44.00%
음성 1회는 취약하다.
N2
N2 → C: 106/145 = 73.10%
N2 → P: 26.90%
음성 2회는 강하지만 확정적이지 않다.
C
C → C: 40/69 = 57.97%
C → P: 42.03%
확인된 국소 음전은 항상 지속되지 않는다.
중단 없는 성공 경로와 최종 음전의 차이
6.82%
중단 없는 단일 성공 경로
지속, 초기화 또는 재시도 없이 P → N1 → N2 → C로 진행.
37.61%
최종적으로 C에 도달
전체 관찰 시도 후 234개 중 88개 시퀀스가 C에 도달.
\(0.1678 \times 0.5564 \times 0.7310 = 0.0682\)
6.82%는 하나의 연속 경로 확률이고, 37.61%는 누적 관찰 종점
비율이다. 반복 시도가 가능하기 때문에 최종 도달 비율이 훨씬 높다.
시간 리본과 상태 점유
20.0일
첫 N1까지의 중앙값
→
38.0일
N2까지의 중앙값
→
48.5일
확인까지의 중앙값
P
관찰 1,906건
N1
300건
N2
153건
C
146건
음전 시작 중앙값 34.5일이 N2 중앙값 38.0일보다 이르게 보이는
것은 두 요약의 대상 시퀀스와 실패한 이전 시도 포함 여부가 다를
수 있기 때문이다. 각 시간 지표의 분모를 별도로 명시할 필요가 있다.
층별 결과의 해석
비교적 유리한 관찰 경로
VRE 소변: P → N1 41.10%; N1 → N2 75.00%; N2 → C 95.00%.
시퀀스 23개로 불확실성을 함께 보아야 한다.
뚜렷한 초기 병목
CRE 대변: P → N1 9.50%; N1 → N2 48.53%; N2 → C 59.38%.
시퀀스 67개로 통합 결과에 큰 영향을 주는 층이다.
희소한 100%
일부 상처 셀의 100% 전이는 한두 건의 사건으로 계산되었다.
안정적 추론이 아니라 기술적 관찰로 유지해야 한다.
IV. 확인 음전 후 재양성
재양성 분모 나무
음전 시퀀스 88개
↓
추적 시퀀스 39개
집계된 음전 후 전이가 최소 1회 존재
↓
재양성 시퀀스 21개
추적 시퀀스의 53.85%
↓
집계된 추적이 없는 시퀀스 49개
전체 음전 시퀀스의 55.68%
↓
재양성 상태를 충분히 관찰할 수 없음
관찰 부재가 지속성의 증거는 아님
하나의 재양성률이 존재하지 않는 이유
분석 단위와 추적 조건이 달라지면 같은 현상의 백분율도 달라진다.
종점
분자 / 분모
추정치
측정 내용
전이 단위 C → P
29 / 69
42.03%
C에서 관찰된 다음 전이 중 양성 비율.
추적 시퀀스 재양성
21 / 39
53.85%
추적된 음전 시퀀스 중 한 번 이상 재양성.
전체 음전 시퀀스 재양성
21 / 88
23.86%
C에 도달한 모든 시퀀스 중 관찰된 재양성.
추적 환자 재양성
17 / 27
62.96%
추적 환자 중 한 시퀀스 이상에서 재양성.
전체 음전 환자 재양성
17 / 43
39.53%
C 시퀀스를 가진 모든 환자 중 관찰된 재양성.
환자 단위 추적 퍼널
음전 환자 43명
→
추적 환자 27명
→
재양성 환자 17명
음전 환자 43명 중 16명은 어떤 음전 시퀀스에서도
집계된 후속 전이가 없었다.
추적 분모에는 감시가 계속된 환자가 선택적으로 많이 포함될 수 있다.
반대로 전체 음전 분모는 관찰되지 않은 경우를 재양성 없음으로 남긴다.
어느 값도 감시 강도와 무관한 환자 발생률이 아니다.
재양성 시점 지도
첫 재양성은 대체로 일찍 발생했지만 301일까지 이어지는 긴 꼬리가 존재한다.
11일
첫 재양성까지의 중앙값
7–31일
사분위 범위
시퀀스 6개
재양성 2회 이상
적절한 재양성 결론
추적이 계속된 상황에서 확인된 국소 음전은 항상 지속되지 않았다.
현재 보고서는 감시 강도와 무관한 환자 재양성 발생률을 추정하지 않는다.
이를 위해서는 검열, 위험 시간, 환자 군집을 포함한 반복 사건 모델이 필요하다.
V. 종점 정렬 말기 분석
종점 코호트 흐름
환자 98명 중 98명 모두 종점 자료와 연계
종점 날짜 충돌 없음
↓
사망 44명
↓
12주 말기 모델 34명
↓
짝지은 환자 28명
이전 창과 말기 창 모두에 존재
↓
검열 종점 54명
↓
12주 말기 모델 43명
↓
짝지은 환자 28명
이전 창과 말기 창 모두에 존재
검열군은 행정적 검열, 퇴원, 전원, 철회를 함께 포함한다. 따라서
생물학적으로 균일한 생존 대조군으로 해석해서는 안 된다.
주별 역시간 경향
마지막 4주를 음영으로 표시하였다. 검열 정렬 음성률은 상승하지만
사망 정렬 경향은 낮은 수준에 머문다.
주별 값은 실제로 채취된 배양만을 설명한다. 채취되지 않았거나
확보할 수 없었던 검체를 음성으로 바꾸지 않았다.
짝지은 말기 창 대비
사망 정렬
이전 창 → 말기 창
↓
−8.53%p
95% 구간: −15.00부터 −2.74%p
짝지은 환자 28명
검열 정렬
이전 창 → 말기 창
↑
+18.29%p
95% 구간: +6.99부터 +28.12%p
짝지은 환자 28명
말기 분석의 핵심은 종점 사이의 대비이다. 사망 정렬 경로에서는
비사망 검열 전과 같은 강한 음성률 상승이 나타나지 않았다.
변화점 근거
사망 정렬 경계
선택 경계: 3주
변화: −4.74%p
변화를 지지하는 ΔBIC: −1.215
부트스트랩 지지율 23.60% · 지지되지 않음
검열 정렬 경계
선택 경계: 3주
변화: +27.37%p
변화를 지지하는 ΔBIC: +13.657
부트스트랩 지지율 75.20% · 강한 탐색적 경계
선택된 경계는 두 구간으로 시기를 요약하는 도구이며, 말기 질환이
생물학적 전환을 유발했다는 근거가 아니다.
세 가지 조정 모델의 관점
혼합효과 로지스틱
환자 무작위 절편을 포함한 선형 4주 경향.
사망 경향 OR: 0.896
사망 대 검열 비: 0.455
환자 CV 로그손실: 0.654
일반화 가법 혼합모델
환자 무작위 절편을 포함한 비선형 곡선.
환자 CV 로그손실: 0.629
개선량: 0.0251
선호 빈도주의 예측 모델
베이지안 계층 모델
라플라스 근사 아래의 확률 진술.
사망 변화: −2.43%p
P(사망 증가): 20.82%
P(사망 변화 > 검열 변화): 0.00%
조정 결과
추정치
해석
검열 종점에 4주 가까워짐
OR 1.968, 95% 구간 1.601–2.420
음성 배양 오즈가 크게 증가하였다.
사망에 4주 가까워짐
OR 0.896, 95% 구간 0.689–1.164
뚜렷한 상승이 없고 방향은 약간 감소하였다.
사망 대 검열 경향 비
OR 0.455, 95% 구간 0.325–0.636
두 종점의 경향이 현저히 달랐다.
채취 강도
OR 1.485, 95% 구간 1.034–2.135
관찰 빈도 자체가 중립적이지 않을 가능성이 있다.
관찰 과정의 한계
임상 상태
→
검체 채취 여부
→
채취 검체 부위
→
관찰된 양성 또는 음성
결측 검체를 양성이나 음성으로 바꾸지 않은 것은 올바르지만 정보성
결측이 사라지는 것은 아니다. 향후에는 검체 채취 확률을 모델링하고,
검열 사유를 분리하며, 검체 부위별 말기 경향을 비교할 필요가 있다.
VI. 경쟁위험 다상태 분석
최초 사건 모델 구조
P
⇄
N1
⇄
N2
확인된 국소 C
흡수 최초 사건
사망
경쟁 흡수 사건
모델은 하나의 환자–병원체–부위 에피소드를 P, N1, N2에서 추적하여
확인된 국소 C 또는 사망 중 먼저 일어나는 사건에서 종료한다.
음전 후 재양성은 이 최초 사건 모델 밖에 있다.
에피소드 종점 구성
C 우선 88개
37.61%
사망 우선 83개
35.47%
검열 63개
26.92%
234
에피소드
91
고유 환자
400 / 400
유효 환자 군집 부트스트랩
에피소드 수와 환자 수는 서로 바꿔 사용할 수 없다. 한 번의 사망이
같은 환자가 제공한 여러 활성 병원체–부위 에피소드를 종료할 수 있다.
전이 강도 지도
현재 상태
100 노출일당 전진율
초기화율
사망률
시각적 의미
P
P → N1: 1.0336
P 유지 자체는 계속되는 노출 시간
P → 사망: 0.2826
지속 양성에서 벗어나는 속도가 느리다.
N1
N1 → N2: 3.4464
N1 → P: 2.8765
N1 → 사망: 0.2442
진행과 초기화가 서로 가깝게 경쟁한다.
N2
N2 → C: 3.7084
N2 → P: 1.3064
N2 → 사망: 0.1686
다음 모델 이탈은 확인 C가 우세하다.
P → N1
100 노출일당 1.0336
N1 → N2
100 노출일당 3.4464
N2 → C
100 노출일당 3.7084
이 값은 배양 1회당 확률이 아니라 노출 시간당 전이율이다.
3일 간격과 14일 간격은 동일한 위험 시간을 제공하지 않는다.
장기 C-선행-사망 확률
음성 근거가 누적될수록 장기 최초 사건의 균형이 C 방향으로 이동한다.
53.40%
P에서 시작
68.00%
N1에서 시작
85.00%
N2에서 시작
시간 지평 지도
고정 시점 확률은 아직 P, N1, N2에 남아 있는 미해결 분율을 보존한다.
P에서 시작
확인 C
사망
P/N1/N2 잔류
30일
2.41%
7.91%
89.68%
90일
16.29%
20.21%
63.50%
180일
32.47%
31.78%
35.75%
365일
47.01%
42.08%
10.91%
“P에서 C가 사망보다 먼저 일어날 장기 확률은 53.40%”와
“P에서 90일 C 확률은 16.29%”는 서로 다른 질문이다. 전자는 장기
흡수확률이고 후자는 고정 시점의 미해결 에피소드를 그대로 남긴다.
모델의 경계
보존되는 요소
불규칙한 노출 시간
경쟁 최초 사건
환자 군집 불확실성
보존되지 않는 요소
정확한 구간 검열 전이 시점
체류 시간에 따른 전이 강도 변화
C 이후 반복 사건
시간에 따라 변하는 감시 강도
VII. HMM 결과와 모델 선택
생물학적 HMM 단계 경사
모든 프로파일에서 단조 감소하지만 감소 크기는 초기 상태 가정에 따라 달라진다.
양성 조건부 1차 프로파일의 평균 잠재 집락 확률은 P 92.53%,
N1 25.50%, N2 2.96%, C 0.94%이다. 방향은 일관되지만 운영 단계와
사후확률이 같은 배양 이력에서 계산되므로 생물학적 완전 제거의
독립 검증은 아니다.
초기 상태 민감도
초기 상태 프로파일
P
N1
N2
C
BIC
양성 조건부 기준선
92.53%
25.50%
2.96%
0.94%
2,206.435
추정 초기 상태
86.54%
24.47%
3.38%
0.63%
2,187.042
정상분포 초기 상태
86.56%
24.67%
3.45%
0.63%
2,179.351
설계에 맞춘 1차 모델
→
대안 BIC가 27.084 개선
→
생물학적 해석은 탐색적으로 유지
핵심 모수와 단계별 사후확률이 의미 있게 달라지고 서로 다른 우도
모드도 두 개 발견되었다. 따라서 근거를 탐색적,
초기 상태 민감도를 매우 민감으로 분류한 판정은 타당하다.
전환 HMM 차수 비교
2체제 모델이 BIC를 최소화하고, 4체제의 추가 예측 개선은 매우 작다.
모델
수렴
AICc
BIC
CV 로그손실
최소 점유율
최소 분리도
4상태 MM / 1체제
예
2,254.700
2,288.967
0.506774
100.00%
NE
2체제
예
2,164.659
2,250.204
0.486243
31.64%
34.13%
3체제
예
2,173.920
2,321.938
0.483468
27.59%
12.57%
4체제
예
2,193.655
2,415.218
0.482243
13.15%
10.74%
5체제
아니오
2,224.257
2,530.284
0.483921
5.15%
12.69%
2체제 전이 영역 지도
체제 2는 전반적으로 진행 영역이 크고, 체제 1은 P 방향 복귀가 우세하다.
체제 1 · 낮은 진행
P → N1: 7.55%
N1 → N2: 28.51%
N2 → C: 42.07%
C → C: 16.46%
관찰 점유율: 68.36%
체제 2 · 높은 진행
P → N1: 54.34%
N1 → N2: 69.77%
N2 → C: 79.30%
C → C: 61.45%
관찰 점유율: 31.64%
두 체제는 전이 패턴을 나타낸다. 이를 “지속 집락 환자”와 “음전 환자”로
명명해서는 안 된다. 점유율과 전환 비대칭은 고정된 환자 유형보다
시퀀스의 단계 또는 시간 경과를 반영할 가능성이 있다.
2체제가 선택된 이유
AICc 최상
+
BIC 최상
+
MM 대비 CV 4.05% 개선
+
허용 가능한 점유율과 분리도
→
선호 모델: 2체제
4체제는 2체제보다 CV 로그손실을 0.0040, 약 0.82%만 개선하지만
BIC는 약 165점 악화되고 체제 분리도도 낮아진다. 모델 차수 안정성을
평가하려면 반복 환자 단위 교차검증이 필요하다.
단순 2상태 계열이 참고 모델로 남아야 하는 이유
전이 동질성
p = 2.536 × 10−21
기억 없음
p = 3.657 × 10−11
마르코프 점검
4개 중 2개 경고
HMM 점검
4개 중 4개 경고
2상태 HMM은 2상태 관찰 마르코프 모델보다 AICc, BIC, 교차검증
로그손실이 좋았다. 그러나 상대적 개선은 절대 적합성을 의미하지
않는다. 따라서 “두 모델 모두 충분하지 않음”이라는 판정은 적절하다.
VIII. 신뢰도 점검과 출력상 문제
문제 우선순위 지도
영향 높음 · 시급성 낮음
반복 교차검증과 외부 HMM 검증
영향 높음 · 시급성 중간
검열 기전 분리와 검체 채취 과정 모델링
영향 높음 · 즉시
자료 조정, 시퀀스 오류 8건, 종점 파서 문제 126건
영향 중간 · 시급성 낮음
보고서 탐색 구조와 접을 수 있는 부록
영향 중간 · 시급성 중간
두 생물학적 HMM 요약의 명칭 분리
영향 중간 · 즉시
0–100% 예측구간과 희소 계수 수정
영향 낮음 · 시급성 낮음
용어와 시각 라벨 정비
영향 낮음 · 시급성 중간
모든 시간 지표 분모의 명시
영향 낮음 · 즉시
재양성 사건표의 세로 막대 이스케이프
우선 수정 항목
우선도
문제
중요한 이유
권장 수정
치명적
50건, 19건, 30건의 관찰 차이가 해결되지 않았다.
성공적 실행만으로 원자료 완전성을 보장할 수 없다.
관찰 단위 조정 원장을 작성한다.
치명적
시퀀스 준비 오류 8건이 남아 있다.
모든 운영 상태와 후속 모델이 시퀀스 구성에 의존한다.
영향 시퀀스, 원본 문장, 규칙, 포함 여부를 표시한다.
치명적
종점 가져오기에 파서 문제 126건이 있다.
말기 및 경쟁위험 모델은 종점 무결성에 의존한다.
심각도와 환자 단위 감사표를 노출한다.
높음
혼합모델 주별 구간이 0–100%이다.
정보성이 없으며 분산 계산 문제일 수 있다.
모집단 신뢰구간, 신규 환자 예측구간, 조건부 구간을 분리한다.
높음
혈액 대 대변 OR가 약 5,260만이다.
혈액 관찰 7건으로 희소자료 또는 분리 문제가 발생한다.
NE 처리, 수준 결합 또는 명시적 벌점 모델을 사용한다.
높음
연계 환자 21명이 말기 모델에서 빠졌으나 흐름표가 없다.
제외 이유를 알 수 없다.
환자별 하나의 명확한 제외 사유를 추가한다.
중간
희소 셀에 100%가 표시된다.
시각적 강조가 확실성으로 오해될 수 있다.
분자, 분모, 추정 가능성 경고를 함께 표시한다.
재양성 사건표 형식 오류
현재 출력
1022|CRE|Stool
Markdown이 세로 막대를 표의 열 구분자로 해석한다.
안전한 출력
1022\|CRE\|Stool
1022 · CRE · Stool
구분자를 이스케이프하거나 표 구분자가 아닌 문자로 바꾸어야 한다.
보고서 구조 개선
현재 독해 부담
긴 기술 문단이 끊기지 않고 이어짐
핵심 결과와 탐색 결과가 같은 층에 배치됨
매우 긴 에피소드 및 시퀀스 표가 본문에 존재
백분율 옆에 분모가 즉시 보이지 않음
→
권장 시각 구조
첫 화면에 1페이지 근거 대시보드
한 섹션에 하나의 시각적 질문
모든 백분율 옆에 분자, 분모, 단위, 추적 조건
긴 감사표는 접을 수 있는 부록으로 이동
희소 또는 불안정 추정치 자동 경고
IX. 연구 결론과 다음 우선순위
주요 강점
프로토콜 충실도
4상태 모델이 3회 음성 규칙을 직접 표현한다.
분모 투명성
전이, 시퀀스, 환자 재양성을 분리한다.
완전한 종점 연계
배양 환자 98명 모두 검증된 종점을 가진다.
군집 고려 불확실성
환자 내 반복 관찰을 분석에 반영한다.
모델 경계 문구
운영적, 생물학적, 인과적, 임상적 의미를 분리한다.
상호보완 분석
기술, 예측, 베이지안, 경쟁위험 질문을 구분한다.
지지되는 실질적 결론
첫 음성
가장 큰 병목
→
N1과 N2
의미 있는 근거 이력
→
세 번째 음성
의미 있는 확인
→
C 이후
지속성은 불확실
사망 정렬 경향은 비사망 검열 정렬 경향과 현저히 다르지만, 역시간
분석과 조정 모델 모두 말기 질환의 인과적 기전을 입증하지 않는다.
후속 작업의 권장 순서
1. 자료 감사를 종결
원자료, 파서, 준비, 종점 문제를 모두 해결한다.
2. 불확실성 출력을 수정
0–100% 구간과 불안정 희소 계수를 바로잡는다.
3. 모델 선택을 안정화
환자 단위 CV를 반복하고 체제 차수 선택을 부트스트랩한다.
4. 검열 기전을 분리
퇴원, 전원, 철회, 행정적 종점을 별도로 분석한다.
5. 검체 채취 과정을 모델링
정보성 채취와 부위별 결측을 평가한다.
6. 반복 사건 모델 개발
위험 시간을 포함한 최초 및 반복 재양성을 추정한다.
7. 다상태 모델 확장
준마르코프와 구간 검열 모형을 검토한다.
8. 외부 검증
HMM 방출확률, 체제, 종점 확률을 다른 코호트에서 검증한다.
논문에 사용할 수 있는 실질적 결론
본 감시 코호트에서 확인된 국소 음전에 이르는 가장 큰 운영적
장벽은 첫 번째 적격 음성 배양을 얻는 단계였다. 음성 근거가 누적될수록
진행 가능성이 높아졌으며, 이는 N1과 N2를 서로 다른 근거 이력
상태로 유지하고 세 번째 음성을 의미 있는 확인 단계로 보존하는
해석을 지지한다. 추적이 계속된 상황에서 확인 음전은 항상 지속되지
않았으나, 재양성 추정치는 분석 단위와 음전 후 추적 가능성에 크게
의존하였다. 사망 정렬 배양에서는 비사망 검열 전의 뚜렷한 음성률
상승과 같은 경향이 나타나지 않았지만, 관찰 연구 설계와 변화하는
검체 확보 가능성 때문에 인과적으로 해석할 수 없다. 4상태 운영
모델은 1차 해석 모델로 유지하고, 2체제 전환 HMM은 2차 예측 층으로
사용할 수 있으며, 생물학적 HMM 모수와 경쟁위험 확률은 명시적으로
모델 기반의 탐색 결과로 남기는 편이 타당하다.
Written on July 30, 2026
Interpreting the updated nGeneMDRO results: what the data actually show (Written July 30, 2026)
Report basis.
This interpretation uses the complete analysis generated on
July 29, 2026 at 22:47 from RawDataset.zip and
nGeneDataset_pattern06.xlsx. The current report contains 29 analysis
sections, 98 patients, 2,703 parsed culture observations, and 2,673 observations
retained after same-day reconciliation.
The most important conclusion is not simply that some cultures became negative.
The principal operational difficulty was obtaining the first qualifying
negative. Once negative evidence accumulated, progression toward confirmed
local clearance became increasingly favorable. However, confirmed clearance was
not always durable, and the present data do not support a terminal
rise in culture negativity before death.
Most next cultures obtained while in P remain positive. The first qualifying negative is the narrowest point.
Primary descriptive result
Does additional negative evidence matter?
N1 → N2: 55.64%; N2 → C: 73.10%
Progression becomes more favorable after each qualifying negative. The third negative still adds confirmation.
Protocol-aligned result
Was clearance always durable?
21 of 39 followed cleared sequences recurred
Recurrence was common when post-clearance surveillance continued.
Follow-up-dependent description
Did negativity rise before death?
No supported increase
The paired change was −8.53 percentage points; the adjusted Bayesian mean was −2.24 points.
Consistent evidence against the proposed increase
What is the 90-day outlook from P?
C 10.59%; Death 23.41%; unresolved 66.00%
Most episodes remain in P, N1, or N2 at 90 days.
Exploratory model-based probability
Which hidden-regime order is preferred?
Two hidden regimes
Two regimes provide the best balance of fit, prediction, occupancy, separation, and parsimony.
Secondary predictive layer
Direct answer to the terminal-negativity hypothesis
Paired 1–4 versus 5–12 weeks
−8.53 pp
95% interval −14.08 to −2.69 pp
Death change-point search
Not supported
Selected week 3; ΔBIC −1.215; bootstrap support 23.60%
Adjusted linear death trend
OR 0.904
95% interval 0.695 to 1.177
Bayesian terminal change
−2.24 pp
P(increase) 22.85%; 95% credible interval −8.19 to +3.58 pp
The hypothesis that culture negativity rises sharply during the final four
or eight weeks before death is not supported by the current result.
The observed direction is more compatible with stable or somewhat lower
negativity near death. This remains an observational statement and does not
prove a terminal biological mechanism.
Each analysis answers a different question
Analysis
Unit
Main quantity
What it must not be mistaken for
Four-state operational model
Patient × pathogen × site sequence
Probability of the next included operational transition
Weekly patient risk
Recurrence analysis
Transition, sequence, or patient
Observed post-C positivity under a stated denominator
One universal recurrence incidence
Reverse-time terminal analysis
Collected culture nested within patient
Negative fraction as an endpoint approaches
Probability that an uncollected specimen was negative
Competing-risk multi-state model
Patient × pathogen × site episode
First confirmed C versus Death
Patient-level mortality or post-C recurrence
Biological HMM
Latent clear/colonized process
Model-based P(colonized)
Externally validated microbiological truth
Switching four-state HMM
Hidden transition regime
Changing transition patterns among P, N1, N2, and C
Fixed patient subtype or biological disease stage
II. Data foundation, provenance, and cohort selection
Dataset composition
CRE contributes approximately two thirds of all parsed observations.
Stool and urine dominate the specimen distribution; blood contains only seven observations.
Domain
Count
Share of 2,703 observations
Interpretive consequence
CRE
1,771
65.52%
Pooled findings are strongly influenced by CRE.
VRE
625
23.12%
Provides the second-largest organism domain.
MRPA + MRAB
307
11.36%
Several organism-site cells remain sparse.
Stool
1,382
51.13%
More than half of observations arise from stool surveillance.
Blood
7
0.26%
Blood coefficients are intrinsically unstable.
Observation reconciliation waterfall
2,772
Manuscript-stated episodes
→
−50
2,722
Manuscript pathogen subtotal
→
−19
2,703
Explicit parser observations
→
−30
2,673
Prepared observations
Difference
Magnitude
Origin
Required action
Manuscript arithmetic gap
50
The stated total does not equal the pathogen subtotal.
Retain as a source-level inconsistency.
Manuscript-to-parser gap
19
Difference between manuscript pathogen counts and explicit reconstructed observations.
Trace to source lines or categories.
Parser-to-preparation reduction
30
Same-day duplicate or conflict reconciliation and sequence preparation.
Provide a deterministic row-level ledger.
These three differences must remain separate. They arise at different
provenance stages and cannot be silently combined into one discrepancy.
Endpoint provenance is the key methodological correction
98 of 98 patients linked to an outcome record
No missing outcome, no extra record, and no endpoint-date conflict
44
Death endpoints
Primary external absorbing endpoint
17
Culture-defined endpoints
Audit and sensitivity only
The number 54 represents all non-death endpoints, but it must
not be called the primary censoring cohort. Seventeen of those endpoints
were generated by the culture trajectory itself. Treating them as neutral
censoring would introduce circularity because the endpoint timing would be
defined by the same cultures being analyzed.
Different questions use different subsets
98 linked culture patients
↓
81 primary endpoint patients
44 Death + 37 independent censoring
↓
60 terminal-model patients
34 Death + 26 independent censoring
↓
77 competing-risk patients
199 eligible episodes
The terminal trend and competing-risk models therefore do not use the same
denominator. This is expected because the models impose different culture,
sequence, and endpoint eligibility requirements.
Audit status
Patient-file parser
5 information notices
0 warnings and 0 errors
Sequence preparation
8 errors
14 additional information notices
Outcome parser
126 information notices
0 warnings and 0 errors
The 126 outcome notices are documented transformations or provenance notices,
not 126 errors. Nevertheless, the eight error-level sequence-preparation
issues and the unresolved 30-observation reduction should be reviewed before
the result is treated as final.
III. Four-state operational clearance
Operational state machine
P
Positive
First qualifying negative 16.78%
→
N1
One qualifying negative
Second qualifying negative 55.64%
→
N2
Two qualifying negatives
Third qualifying negative 73.10%
→
C
Confirmed local clearance
N1 → P: 44.00%
One negative is easily reset by a later positive.
N2 → P: 26.90%
Two negatives are strong evidence but remain incomplete.
C → P: 42.03%
A later positive may occur after local clearance.
Each qualifying negative must be at least three days after the preceding
qualifying negative. A positive culture before C resets the run to P.
Local C is not the highest clearance level
Local C
One patient × one pathogen × one site
→
PLC
All required sites for one pathogen
→
PtLC
All required pathogen-site units
The current four-state analysis is primarily a local
patient–pathogen–site model. Reaching C in one sequence must not be
generalized automatically to pathogen-level or patient-level clearance.
Transition bottleneck profile
Forward progression becomes more favorable after negative evidence begins to accumulate.
Transition
Count / denominator
Probability
Patient-clustered 95% interval
Meaning
P → N1
299 / 1,782
16.78%
14.09%–19.82%
Starts a qualifying negative run.
N1 → N2
153 / 275
55.64%
49.34%–61.31%
One negative progresses to two.
N2 → C
106 / 145
73.10%
66.13%–80.84%
The third qualifying negative confirms C.
C → P
29 / 69
42.03%
31.37%–52.48%
A later positive follows local C.
Cumulative reach funnel
234 eligible sequences
All begin in P
↓
144 reached N1
61.54% of eligible sequences
↓
108 reached N2
46.15% of eligible sequences
↓
88 reached C
37.61% of eligible sequences
These percentages describe whether a sequence ever reached each stage during
follow-up. They are not the same as the probability of the next transition.
Why 6.82% and 37.61% are both correct
6.82%
One uninterrupted path
P → N1 → N2 → C without persistence, reset, or another attempt.
37.61%
Eventually reached C
88 of 234 sequences reached C after all observed attempts.
\(0.1678 \times 0.5564 \times 0.7310 = 0.0682\)
Repeated attempts explain the difference. The first value is a path
probability; the second is a cumulative observed endpoint fraction.
Operational stage and latent-colonization gradient
Every HMM specification shows a steep P-to-C decline, but the numerical level depends on the model and initial-state assumption.
HMM summary
P
N1
N2
C
Interpretive status
Stratified posterior linkage
89.14%
13.23%
1.22%
0.29%
Supported with caution
Positive-conditioned primary profile
92.53%
25.50%
2.96%
0.94%
Exploratory
Estimated initial-state profile
86.54%
24.47%
3.38%
0.63%
Sensitivity analysis
Stationary initial-state profile
86.56%
24.67%
3.45%
0.63%
BIC-preferred sensitivity profile
The monotonic gradient is coherent, but it is not an independent validation
of biological eradication because both the operational stages and HMM
posteriors are derived from the same culture history.
Pathogen–site domain coverage map
Bubble size represents the number of eligible sequences. Color represents the fraction reaching C.
Stratum
Sequences
Reached C
Fraction reaching C
P → N1
N2 → C
C → P
CRE · Sputum
31
8
25.81%
15.71%
62.50%
44.44%
CRE · Stool
67
16
23.88%
9.50%
59.38%
42.86%
CRE · Urine
35
14
40.00%
18.75%
74.07%
47.06%
MRAB · Sputum
14
9
64.29%
67.86%
90.91%
66.67%
MRPA · Urine
10
5
50.00%
32.50%
80.00%
50.00%
VRE · Stool
36
13
36.11%
15.56%
65.00%
14.29%
VRE · Urine
23
16
69.57%
41.10%
95.00%
33.33%
CRE stool is the largest stratum and has the lowest P → N1 estimate among
the large groups. VRE urine appears more favorable, but organism-site
comparisons remain descriptive and require uncertainty-aware modeling.
Apparent 100% values in very small wound cells are not stable estimates.
IV. Recurrence after confirmed local clearance
The denominator tree
88 cleared sequences
↓
39 followed sequences
At least one counted post-C transition
↓
21 recurrent sequences
53.85% of followed sequences
↓
49 sequences without counted follow-up
55.68% of all cleared sequences
↓
Durability not equally observable
No observation is not proof of no recurrence
There is no single recurrence percentage
The estimated percentage changes because the statistical unit and follow-up requirement change.
Endpoint
Numerator / denominator
Estimate
95% Wilson interval
Meaning
Transition-level C → P
29 / 69
42.03%
31.11%–53.79%
Positive next transition among counted transitions out of C.
Followed cleared sequences
21 / 39
53.85%
38.57%–68.43%
At least one recurrence among sequences with follow-up.
All cleared sequences
21 / 88
23.86%
16.17%–33.74%
Observed recurrence among all sequences reaching C.
Followed cleared patients
17 / 27
62.96%
44.23%–78.47%
At least one recurrent sequence among followed patients.
All cleared patients
17 / 43
39.53%
26.37%–54.42%
Observed recurrence among all patients with at least one C sequence.
Recurrence timing is strongly right-skewed
Most recurrences cluster early, but the long tail reaches 301 days.
11 days
Median time to first recurrence
7–31 days
Interquartile range
6 sequences
More than one recurrence
301 days
Longest listed interval
Stratum-level recurrence
Stratum
Cleared
Followed
Recurrent
Recurrence among followed
Median days
CRE · Sputum
8
4
3
75.00%
8.0
CRE · Stool
16
9
5
55.56%
27.0
CRE · Urine
14
8
4
50.00%
7.0
MRAB · Sputum
9
2
1
50.00%
7.0
MRPA · Urine
5
3
2
66.67%
67.0
VRE · Stool
13
5
1
20.00%
7.0
VRE · Urine
16
7
4
57.14%
29.5
Values such as 100% in MRAB wound are based on one followed sequence and one
event. Such cells are useful as event descriptions but are not stable
organism-site recurrence estimates.
Appropriate recurrence conclusion
Confirmed local clearance was not uniformly durable when surveillance
continued. The present report does not estimate a surveillance-independent
patient recurrence incidence. A recurrent-event analysis with censoring,
time at risk, and patient clustering is required for that purpose.
V. Terminal hypothesis and endpoint-aligned trajectories
Observed reverse-time trajectory
Point size reflects the number of patients represented during the week. Vertical bars are patient-clustered 95% intervals.
The death-aligned weekly fraction remains generally low and does not display
a sustained rise during weeks 1–4. The independent-censoring trajectory is
variable and reaches 44.93% in the final week, but fewer patients contribute
to several earlier censoring weeks.
Four-week comparison: aggregate and paired estimates are not the same
These bars use aggregate patient-weighted fractions. The paired mean difference below uses only patients represented in both windows.
Death-aligned paired comparison
−8.53 pp
28 paired patients
95% interval −14.08 to −2.69 pp
Independent-censoring paired comparison
−2.05 pp
15 paired patients
95% interval −15.07 to +9.53 pp
In the independent-censoring cohort, the aggregate patient-weighted fraction
rises from 30.34% to 36.77%, while the mean within-patient paired change is
−2.05 points. This is not an arithmetic contradiction. The aggregate and
paired summaries use different patient sets and weighting rules; only 15
censoring patients are represented in both windows.
No stable four-week or eight-week death boundary was found
Positive values indicate a higher terminal fraction; negative values indicate a lower terminal fraction.
Cohort
Selected boundary
Difference
ΔBIC favoring change
Bootstrap support
Evidence
Death
Week 3
−4.74 pp
−1.215
23.60%
Not supported
Independent censoring
Week 2
+10.59 pp
−2.115
37.80%
Not supported
The death boundary varies broadly from 3 to 10 weeks across bootstrap
samples, and its difference interval spans both negative and positive values.
Therefore, the present data do not identify a reproducible death-related
switch at either four weeks or eight weeks.
Adjusted linear trend
Four weeks closer to independent censoring
OR 1.426
95% interval 1.086 to 1.873
Four weeks closer to death
OR 0.904
95% interval 0.695 to 1.177
Death-versus-censoring trend ratio
OR 0.634
95% interval 0.434 to 0.927
Sampling intensity
OR 1.826
95% interval 1.212 to 2.752
The interaction is more informative than the death slope alone. Endpoint
proximity behaves differently in the two cohorts. Sampling intensity is also
associated with negativity, indicating that the observation process may not
be neutral.
Prediction versus parsimony: why GAMM was selected
Mixed-effects logistic
14 fixed effects
AICc 1026.204
BIC 1099.624
Patient CV log loss 0.645675
Wins on simplicity and information criteria
⇄
Generalized additive mixed model
24 fixed effects
AICc 1031.765
BIC 1153.620
Patient CV log loss 0.622606
Selected by the configured predictive-improvement rule
The GAMM lowers patient-level cross-validation log loss by 0.0231,
approximately 3.6% relative to the linear mixed model. AICc and BIC favor
the simpler model, while out-of-patient prediction favors the nonlinear
curve. The GAMM should therefore be described as the preferred
prediction model, not as proof of a nonlinear biological mechanism.
Adjusted Bayesian trajectories
Shaded areas are 95% credible intervals under the Laplace-approximation posterior.
Posterior contrast
Mean change
95% credible interval
Probability of an increase
Death terminal minus earlier
−2.24 pp
−8.19 to +3.58 pp
22.85%
Independent-censoring terminal minus earlier
+9.19 pp
+1.74 to +16.49 pp
99.33%
Death change minus censoring change
−11.44 pp
−20.99 to −1.76 pp
P(death change > censoring change) = 1.10%
The paired independent-censoring estimate and the adjusted Bayesian estimate
differ because they answer different questions. The paired analysis uses
only 15 patients represented in both windows. The adjusted model uses 1,019
cultures from 60 patients, adjusts for covariates, and applies hierarchical
shrinkage. The robust cross-model conclusion is that the death trajectory
does not show a terminal rise comparable with independent censoring.
What the terminal result does not prove
Supported statement
Collected cultures near death do not show a supported increase in negativity.
Unsupported statement
Terminal illness causes persistent positivity or prevents biological clearance.
Remaining observation bias
Specimen availability, sampling intensity, and sampled sites may change near an endpoint.
VI. Competing-risk P / N1 / N2 / C / Death model
Cohort and first-event outcomes
199
Included episodes
77
Unique patients
54
Confirmed-C first
83
Death first
62
Right censored
C first
27.14%
Death first
41.71%
Right censored
31.16%
These are crude episode outcomes. They are not the same as eventual
absorption probabilities because nearly one third of episodes are censored.
First-event state structure
P
⇄
N1
⇄
N2
Confirmed local C
Absorbing first event
Death
Competing absorbing event
Post-C recurrence is intentionally outside this first-event model. A
non-death independent endpoint is right censoring.
Transition intensities retain irregular observation time
Rates are calculated per 100 exposure-days in the source state, not per culture.
Source state
Forward rate
Reset rate
Death rate
Interpretation
P
P → N1: 0.8612
Remaining P is continued exposure
0.3224
Leaving persistent positivity is slow.
N1
N1 → N2: 2.7965
N1 → P: 2.4751
0.2893
Progression and reset are closely balanced.
N2
N2 → C: 2.5776
N2 → P: 1.1933
0.1909
Confirmed C is the dominant next exit.
Eventual C before Death depends strongly on the current state
Accumulated negative evidence moves the eventual first-event balance toward C.
Starting state
C before Death
95% interval
Death before C
P
42.07%
30.29%–52.89%
57.93%
N1
57.81%
45.77%–68.52%
42.19%
N2
77.73%
66.65%–85.57%
22.27%
Fixed-horizon probabilities from P
Fixed-horizon estimates preserve the large unresolved P/N1/N2 fraction.
Horizon from P
Confirmed C
Death
Still P/N1/N2
30 days
1.32%
9.05%
89.63%
60 days
5.51%
16.83%
77.67%
90 days
10.59%
23.41%
66.00%
180 days
23.22%
37.52%
39.26%
365 days
35.67%
51.01%
13.32%
The eventual P-state C-before-Death probability of 42.07% is not the same as
the 90-day C probability of 10.59%. The eventual quantity continues the
fitted process until absorption; the 90-day result leaves 66.00% unresolved.
Model boundary
What is retained
Irregular time at risk
Competing first events
Patient-cluster bootstrap uncertainty
Explicit same-day tie rule
What is not modeled directly
Exact interval-censored transition times
Duration-dependent transition rates
Post-C recurrent events
Changing surveillance intensity
VII. Biological HMM and switching-HMM interpretation
Biological HMM: coherent gradient but high initial-state sensitivity
Design-aligned primary profile
BIC 2206.435
Fixes baseline hidden state to Colonized after positive enrollment.
Estimated initial state
BIC 2187.042
Improves BIC by 19.392 points relative to primary.
Stationary initial state
BIC 2179.351
BIC-preferred profile; improvement 27.084 points.
All profiles converge and preserve a monotonic P-to-C gradient, but the
baseline hidden-state assumption materially changes fit, parameters, and
stage posteriors. The biological HMM must therefore remain
exploratory.
Switching-HMM order comparison
Lower BIC and lower CV log loss are preferable. The five-regime model did not converge.
Model
Converged
AICc
BIC
CV log loss
Minimum occupancy
Minimum separation
Four-state MM / one regime
Yes
2254.700
2288.967
0.506774
100.00%
NE
Two regimes
Yes
2164.659
2250.204
0.486243
31.64%
34.13%
Three regimes
Yes
2173.920
2321.938
0.483468
27.59%
12.57%
Four regimes
Yes
2193.655
2415.218
0.482243
13.15%
10.74%
Five regimes
No
2224.257
2530.284
0.483921
5.15%
12.69%
The four-regime model improves CV log loss over the two-regime model by only
0.0040, approximately 0.82%, while worsening BIC by about 165 points and
reducing transition-profile separation. Two regimes are therefore the more
defensible compromise.
What the two regimes look like
Regime 2 has a substantially larger progression domain than Regime 1.
Feature
Regime 1
Regime 2
P → N1
7.55%
54.34%
N1 → N2
28.51%
69.77%
N2 → C
42.07%
79.30%
C → C retention
16.46%
61.45%
Observed occupancy
68.36%
31.64%
Stationary probability
10.56%
89.44%
Regime 1 is a low-progression, high-return-to-P pattern. Regime 2 is a
high-progression, more clearance-retentive pattern. These labels describe
transition dynamics and must not be converted automatically into fixed
patient phenotypes.
Why the simple two-state models remain only a reference
Transition homogeneity
p = 2.536 × 10−21
A single pooled process is not supported.
Memorylessness
p = 3.657 × 10−11
The current binary result is insufficient history.
Markov predictive checks
2 of 4 flagged
Absolute adequacy is limited.
Two-state HMM checks
4 of 4 flagged
Relative improvement does not restore adequacy.
The two-state HMM improves AICc by 70.642 points, BIC by 58.932 points,
and CV log loss by 2.53% relative to the two-state Markov model. Nevertheless,
both models fail important structural or predictive checks. “Neither model
adequate” is therefore the correct conclusion.
VIII. Concrete sequence example and reliability review
Worked example: patient 1001, CRE stool
Every observed culture was positive. The sequence remained in P throughout.
Date
Observed culture
Gap
Operational state
HMM P(colonized)
2024-02-20
Positive
Baseline
P → P
99.14%
2024-02-27
Positive
7 days
P → P
99.52%
2024-03-04
Positive
6 days
P → P
99.61%
2024-03-07
Positive
3 days
P → P
99.60%
2024-03-14
Positive
7 days
P → P
99.47%
2024-03-21
Positive
7 days
P → P
98.96%
2024-03-26
Positive
5 days
P → P
97.00%
This sequence illustrates the distinction between observed and latent
quantities. The operational state remains P because every culture is
positive. The HMM posterior changes slightly because it reflects the fitted
latent process, but it remains very high and does not imply clearance.
Specific issues requiring attention
Priority
Issue
Why it matters
Recommended correction
Critical
Fifty-, nineteen-, and thirty-observation gaps remain at different provenance stages.
Successful execution does not establish source-data completeness.
Create an observation-level reconciliation ledger.
Every four-state and downstream model depends on reconstructed sequences.
List the affected sequence, source line, rule, and inclusion decision.
High
Mixed-model weekly intervals are displayed as 0%–100%.
These intervals are non-informative and may reflect an incorrect prediction-variance target.
Separate population confidence, new-patient prediction, and conditional intervals.
High
Blood-versus-stool OR is approximately 51.6 million with SE 2,308.8.
Only seven blood observations exist, producing sparse-data or separation instability.
Suppress as NE, combine unsupported levels, or use a documented penalized model.
Moderate
Independent-censoring paired analysis contains only 15 patients.
The paired terminal estimate is imprecise and sensitive to cohort composition.
Retain the denominator beside every estimate.
Moderate
Repeated patient-level CV has not been run for switching-HMM order.
One fold assignment may not establish order stability.
Repeat folds across seeds and summarize selection frequency.
Moderate
Several sparse strata display 100% transitions.
Visual prominence may be mistaken for certainty.
Display numerator, denominator, and estimability warnings.
Formatting
Sequence identifiers such as 1022|CRE|Stool break Markdown tables.
Vertical bars are interpreted as column delimiters.
Use 1022\|CRE\|Stool or 1022 · CRE · Stool.
Evidence-strength map
More directly supported
Four-state transition counts
Endpoint linkage and provenance
Recurrence denominators
Observed terminal weekly fractions
Supported with caution
Adjusted mixed and GAMM trends
Two-regime switching HMM
Patient-cluster bootstrap intervals
Descriptive but follow-up dependent
Post-C recurrence
Sparse pathogen-site contrasts
Terminal paired-window estimates
Exploratory or not supported
Biological HMM parameters
Terminal change point
Competing-risk absorption probabilities
Simple two-state adequacy
IX. Final interpretation and next priorities
What may reasonably be concluded
Conclusion
Current assessment
The first qualifying negative is the main operational bottleneck.
Supported descriptively
N1 and N2 contain distinct evidence histories and should remain separate.
Supported descriptively
The third negative remains a meaningful confirmation step.
Supported descriptively
Confirmed local clearance may be followed by later positivity.
Supported under continued surveillance
Culture negativity rises before death.
Not supported
Two hidden transition regimes improve prediction over the single-regime MM.
Supported with caution
The hidden regimes are fixed biological patient types.
Not established
Local C proves biological eradication or permits isolation release.
Not established
Recommended order of further work
Close the data audit.
Resolve the manuscript arithmetic gap, the parser gap, the 30-observation
preparation reduction, and all eight sequence-preparation errors.
Repair uncertainty displays.
Correct the 0%–100% mixed-model intervals and suppress unstable sparse
coefficients such as Blood versus Stool.
Repeat patient-level validation.
Re-run cross-validation across multiple fold assignments and summarize
how often two, three, or four regimes are selected.
Model the observation process.
Examine whether specimen collection, sampled site, and sampling intensity
change near Death or independent censoring.
Develop recurrent-event analysis.
Estimate first and repeated recurrence using time at risk, censoring, and
patient clustering.
Extend the multi-state model.
Evaluate semi-Markov or interval-censored models in which rates may depend
on time already spent in P, N1, or N2.
Seek external validation.
Biological HMM emissions, hidden regimes, and model-based C-before-Death
probabilities should remain exploratory until replicated.
Manuscript-ready substantive conclusion
In this long-term care surveillance cohort, the principal operational barrier
to confirmed local clearance was obtaining the first qualifying negative
culture. Once negative evidence accumulated, forward progression became
increasingly favorable, supporting retention of N1 and N2 as distinct
evidence-history states and preserving the third qualifying negative as a
meaningful confirmation step. Confirmed local clearance was not uniformly
durable under continued surveillance, although recurrence estimates depended
substantially on the unit of analysis and availability of post-clearance
follow-up. After culture-defined endpoints were separated from independent
censoring endpoints, death-aligned cultures showed no supported terminal
increase in negativity. The four-state operational model should remain the
primary interpretable analysis. The two-regime switching HMM may serve as a
secondary predictive model, whereas biological HMM parameters, change-point
estimates, and competing-risk absorption probabilities should remain
explicitly exploratory and should not be used as isolation-release or
treatment thresholds.
업데이트된 nGeneMDRO 분석 결과의 구체적 해석
분석 기준.
본 해석은 RawDataset.zip과 nGeneDataset_pattern06.xlsx에서
2026년 7월 29일 22시 47분에 생성된 전체 결과를 기준으로 한다.
현재 보고서에는 분석 섹션 29개, 환자 98명, 파싱된 배양 관찰 2,703건,
동일 날짜 조정 후 유지된 관찰 2,673건이 포함되어 있다.
핵심은 단순히 일부 배양이 음성이 되었다는 사실이 아니다.
확인된 국소 음전으로 가는 과정에서 가장 어려운 단계는
첫 번째 적격 음성을 얻는 것이었다. 음성 근거가 누적된
이후에는 확인 음전 방향으로 진행할 가능성이 높아졌다. 그러나 확인 음전이
항상 지속되지는 않았으며, 현재 자료는 사망 전에 배양 음성률이 갑자기
상승한다는 가설을 지지하지 않는다.
I. 결과를 한눈에 보는 지도
핵심 대시보드
98
배양 환자
2,703
파싱 관찰
234
4상태 시퀀스
88
C 도달 시퀀스
16.78%
P → N1 병목
2체제
선호 전환 HMM
배양 기록
양성 또는 음성 관찰
→
운영 이력
P → N1 → N2 → C
→
지속성
C → P 재양성
→
환자 종점
사망 또는 독립 검열
→
통계 모델
다상태, GAMM, 베이지안, HMM
가장 중요한 여섯 가지 결론
질문
현재 결과
구체적 의미
근거 역할
어느 단계가 가장 어려운가?
P → N1: 16.78%
P에서 시행된 다음 배양의 대부분은 다시 양성이었다. 첫 적격 음성 단계가 가장 좁은 병목이다.
1차 기술 결과
음성 근거가 누적될수록 의미가 커지는가?
N1 → N2: 55.64%; N2 → C: 73.10%
적격 음성이 추가될수록 확인 음전 방향의 진행이 유리해진다.
프로토콜 대응 결과
확인 음전은 항상 유지되었는가?
추적된 음전 시퀀스 39개 중 21개 재양성
음전 후 감시가 계속된 경우 재양성이 흔하였다.
추적 의존 기술 결과
사망 전에 음성률이 상승했는가?
지지되는 상승 없음
짝지은 변화는 −8.53%p였고, 조정 베이지안 평균은 −2.24%p였다.
제안된 상승 가설에 반하는 일관된 결과
P에서 시작한 90일 전망은?
C 10.59%; 사망 23.41%; 미해결 66.00%
대부분은 90일에도 P, N1 또는 N2에 남는다.
탐색적 모델 확률
숨은 체제 수는 몇 개가 적절한가?
2개 숨은 체제
적합도, 예측력, 점유율, 분리도, 단순성의 균형이 가장 좋다.
2차 예측 층
임종 전 음성률 상승 가설에 대한 직접 답변
1–4주 대 5–12주 짝지은 비교
−8.53%p
95% 구간 −14.08부터 −2.69%p
사망 변화점 탐색
지지되지 않음
선택 3주; ΔBIC −1.215; 부트스트랩 지지율 23.60%
조정 선형 사망 경향
OR 0.904
95% 구간 0.695–1.177
베이지안 말기 변화
−2.24%p
증가 확률 22.85%; 95% 신용구간 −8.19부터 +3.58%p
사망 전 4주 또는 8주 이내에 배양 음성률이 갑자기 상승한다는 가설은
현재 결과에서 지지되지 않는다. 관찰된 방향은 사망에
가까워질수록 음성률이 유지되거나 다소 낮아지는 경향에 더 가깝다.
다만 이는 관찰 결과이며 말기 생물학적 기전을 인과적으로 입증하지 않는다.
분석마다 서로 다른 질문에 답한다
분석
분석 단위
주요 추정량
혼동해서는 안 되는 것
4상태 운영 모델
환자 × 병원체 × 부위 시퀀스
다음 포함 배양에서의 운영 상태 전이확률
주 단위 환자 위험
재양성 분석
전이, 시퀀스 또는 환자
명시된 분모 아래 C 이후 관찰된 양성
하나의 보편적 재양성 발생률
역시간 말기 분석
환자 안에 반복된 실제 채취 배양
종점이 가까워질 때 음성 분율
채취되지 않은 검체의 음성 확률
경쟁위험 다상태 모델
환자 × 병원체 × 부위 에피소드
확인 C와 사망 중 최초 사건
환자 단위 사망률 또는 C 이후 재양성
생물학적 HMM
잠재 Clear/Colonized 과정
모델 기반 P(colonized)
외부에서 검증된 미생물학적 진실
전환 4상태 HMM
숨은 전이 체제
P, N1, N2, C 전이 패턴의 변화
고정된 환자 아형 또는 생물학적 단계
II. 자료 기반, 종점 출처, 코호트 선택
자료 구성
CRE가 전체 파싱 관찰의 약 3분의 2를 차지한다.
대변과 소변이 대부분을 차지하며 혈액 관찰은 7건뿐이다.
영역
관찰 수
2,703건 중 비율
해석상 의미
CRE
1,771
65.52%
통합 결과가 CRE에 크게 좌우된다.
VRE
625
23.12%
두 번째로 큰 병원체 영역이다.
MRPA + MRAB
307
11.36%
여러 병원체–부위 셀이 희소하다.
대변
1,382
51.13%
전체 관찰의 절반 이상이 대변 감시에서 나온다.
혈액
7
0.26%
혈액 관련 회귀계수는 본질적으로 불안정하다.
관찰 수 조정 폭포도
2,772
원고 기재 전체 배양
→
−50
2,722
원고 병원체별 소계
→
−19
2,703
명시적 파서 관찰
→
−30
2,673
준비된 관찰
차이
크기
발생 단계
필요한 조치
원고 산술 차이
50
원고의 전체 수와 병원체별 소계가 일치하지 않는다.
원자료 불일치로 별도 유지한다.
원고–파서 차이
19
원고 병원체 수와 명시적 재구성 관찰의 차이이다.
원본 문장 또는 범주까지 추적한다.
파서–준비 감소
30
동일 날짜 중복·충돌 조정과 시퀀스 준비 과정이다.
관찰 단위 결정 원장을 제공한다.
세 차이는 서로 다른 출처 단계에서 발생한다. 하나의 총차이로 합치면
어느 단계에서 수정해야 하는지 알 수 없게 된다.
종점 출처 분리가 가장 중요한 방법론적 개선이다
환자 98명 중 98명 모두 종점 기록과 연결
종점 누락, 코호트 외 기록, 종점 날짜 충돌 없음
44명
사망 종점
외부 흡수 종점
37명
독립 비사망 검열
1차 비사망 비교군
17명
배양 정의 종점
감사 및 민감도 분석만 사용
비사망 종점 전체는 54명이지만, 이를 1차 검열군이라고 부르면 안 된다.
그중 17명의 종점 시점은 배양 경로 자체로 정의되었다. 이를 중립적인
검열로 사용하면 분석 대상인 배양 결과가 다시 종점 시점을 결정하는
순환성이 생긴다.
질문에 따라 사용되는 하위 코호트가 다르다
종점 연결 환자 98명
↓
1차 종점 환자 81명
사망 44명 + 독립 검열 37명
↓
말기 모델 환자 60명
사망 34명 + 독립 검열 26명
↓
경쟁위험 환자 77명
적격 에피소드 199개
말기 경향 모델과 경쟁위험 모델의 분모가 다른 것은 오류가 아니다.
두 모델이 요구하는 배양, 시퀀스, 종점 적격 조건이 다르기 때문이다.
자료 감사 상태
환자 파일 파서
정보 5건
경고 0건, 오류 0건
시퀀스 준비
오류 8건
추가 정보 14건
종점 파서
정보 126건
경고 0건, 오류 0건
종점 파서의 126건은 문서화된 변환 또는 출처 알림이며 오류 126건이
아니다. 그러나 시퀀스 준비 오류 8건과 30건의 관찰 감소는 최종
판정 전에 반드시 검토할 필요가 있다.
III. 4상태 운영적 음전
운영 상태 기계
P
양성
첫 적격 음성 16.78%
→
N1
적격 음성 1회
두 번째 적격 음성 55.64%
→
N2
적격 음성 2회
세 번째 적격 음성 73.10%
→
C
확인된 국소 음전
N1 → P: 44.00%
음성 1회는 이후 양성에 의해 쉽게 초기화된다.
N2 → P: 26.90%
음성 2회는 강한 근거지만 아직 완전하지 않다.
C → P: 42.03%
국소 음전 이후에도 양성이 다시 나타날 수 있다.
각 적격 음성은 이전 적격 음성과 최소 3일 이상 떨어져 있어야 한다.
C에 도달하기 전 양성이 나오면 P로 초기화된다.
국소 C는 최상위 음전 단계가 아니다
국소 C
환자 1명 × 병원체 1종 × 부위 1곳
→
PLC
한 병원체의 모든 필요 부위 음전
→
PtLC
모든 필요 병원체–부위 단위 음전
현재 4상태 분석은 주로 환자–병원체–부위 단위의
국소 모델이다. 한 시퀀스가 C에 도달했다는 사실을
병원체 전체 또는 환자 전체 음전으로 자동 확대해서는 안 된다.
전이 병목 구조
음성 근거가 누적되기 시작하면 다음 단계로 전진할 가능성이 높아진다.
전이
사건 수 / 분모
확률
환자 군집 95% 구간
의미
P → N1
299 / 1,782
16.78%
14.09%–19.82%
적격 음성 연속 과정을 시작한다.
N1 → N2
153 / 275
55.64%
49.34%–61.31%
음성 1회가 2회로 진행한다.
N2 → C
106 / 145
73.10%
66.13%–80.84%
세 번째 적격 음성이 C를 확인한다.
C → P
29 / 69
42.03%
31.37%–52.48%
국소 C 이후 양성이 나타난다.
누적 도달 퍼널
적격 시퀀스 234개
모두 P에서 시작
↓
N1 도달 144개
적격 시퀀스의 61.54%
↓
N2 도달 108개
적격 시퀀스의 46.15%
↓
C 도달 88개
적격 시퀀스의 37.61%
이 비율은 추적 기간 중 각 단계에 한 번이라도 도달했는지를 나타낸다.
다음 한 번의 전이확률과는 다른 값이다.
6.82%와 37.61%가 모두 맞는 이유
6.82%
중단 없는 단일 경로
지속, 초기화, 재시도 없이 P → N1 → N2 → C로 진행.
37.61%
최종적으로 C 도달
전체 관찰 시도 후 234개 중 88개 시퀀스가 C에 도달.
\(0.1678 \times 0.5564 \times 0.7310 = 0.0682\)
반복 시도가 허용되므로 두 값이 달라진다. 첫 값은 경로 확률이고,
두 번째 값은 누적 관찰 종점 비율이다.
운영 단계와 잠재 집락 확률
모든 HMM 사양에서 P에서 C로 급격히 감소하지만 수치 수준은 모델과 초기 상태 가정에 따라 달라진다.
HMM 요약
P
N1
N2
C
해석 상태
층화 사후연계
89.14%
13.23%
1.22%
0.29%
주의를 동반한 지지
양성 조건부 1차 프로파일
92.53%
25.50%
2.96%
0.94%
탐색적
추정 초기 상태 프로파일
86.54%
24.47%
3.38%
0.63%
민감도 분석
정상분포 초기 상태 프로파일
86.56%
24.67%
3.45%
0.63%
BIC 선호 민감도 프로파일
단조 감소는 방향적으로 일관되지만 생물학적 완전 제거의 독립 검증은
아니다. 운영 단계와 HMM 사후확률 모두 같은 배양 이력에서 계산되기 때문이다.
병원체–검체 부위 영역 지도
버블 크기는 적격 시퀀스 수, 색상은 C에 도달한 시퀀스 비율을 나타낸다.
층
시퀀스
C 도달
C 도달 비율
P → N1
N2 → C
C → P
CRE · 객담
31
8
25.81%
15.71%
62.50%
44.44%
CRE · 대변
67
16
23.88%
9.50%
59.38%
42.86%
CRE · 소변
35
14
40.00%
18.75%
74.07%
47.06%
MRAB · 객담
14
9
64.29%
67.86%
90.91%
66.67%
MRPA · 소변
10
5
50.00%
32.50%
80.00%
50.00%
VRE · 대변
36
13
36.11%
15.56%
65.00%
14.29%
VRE · 소변
23
16
69.57%
41.10%
95.00%
33.33%
CRE 대변은 가장 큰 층이며 대규모 층 가운데 P → N1이 가장 낮다.
VRE 소변은 비교적 유리해 보이지만 병원체–부위 비교는 아직 기술적
결과이다. 작은 상처 셀의 100%는 안정적인 우수성을 뜻하지 않는다.
IV. 확인된 국소 음전 후 재양성
재양성 분모 나무
음전 시퀀스 88개
↓
추적 시퀀스 39개
C 이후 집계된 전이가 최소 1회 존재
↓
재양성 시퀀스 21개
추적 시퀀스의 53.85%
↓
집계된 추적이 없는 시퀀스 49개
전체 음전 시퀀스의 55.68%
↓
지속성을 동일하게 관찰할 수 없음
관찰이 없다는 사실은 재양성이 없다는 증거가 아님
하나의 재양성 백분율은 존재하지 않는다
통계 단위와 추적 조건이 달라지므로 같은 현상의 백분율도 달라진다.
종점
분자 / 분모
추정치
95% Wilson 구간
의미
전이 단위 C → P
29 / 69
42.03%
31.11%–53.79%
C에서 집계된 다음 전이 중 양성 비율.
추적된 음전 시퀀스
21 / 39
53.85%
38.57%–68.43%
추적 시퀀스 중 한 번 이상 재양성.
전체 음전 시퀀스
21 / 88
23.86%
16.17%–33.74%
C에 도달한 모든 시퀀스 중 관찰된 재양성.
추적된 음전 환자
17 / 27
62.96%
44.23%–78.47%
추적 환자 중 한 시퀀스 이상 재양성.
전체 음전 환자
17 / 43
39.53%
26.37%–54.42%
C 시퀀스를 가진 모든 환자 중 관찰된 재양성.
재양성 시간은 오른쪽으로 긴 분포를 가진다
대부분의 재양성은 조기에 모이지만 긴 꼬리는 301일까지 이어진다.
11일
첫 재양성까지의 중앙값
7–31일
사분위 범위
시퀀스 6개
재양성 2회 이상
301일
가장 긴 보고 간격
층별 재양성
층
음전
추적
재양성
추적 시퀀스 중 재양성
중앙값 일수
CRE · 객담
8
4
3
75.00%
8.0
CRE · 대변
16
9
5
55.56%
27.0
CRE · 소변
14
8
4
50.00%
7.0
MRAB · 객담
9
2
1
50.00%
7.0
MRPA · 소변
5
3
2
66.67%
67.0
VRE · 대변
13
5
1
20.00%
7.0
VRE · 소변
16
7
4
57.14%
29.5
MRAB 상처의 100%와 같은 값은 추적 시퀀스 1개와 사건 1개에 근거한다.
사건 설명에는 유용하지만 안정적인 병원체–부위 재양성률은 아니다.
적절한 재양성 결론
감시가 계속된 상황에서 확인된 국소 음전은 항상 유지되지 않았다.
현재 보고서는 감시 강도와 무관한 환자 재양성 발생률을 추정하지 않는다.
이를 위해서는 검열, 위험 시간, 환자 군집을 포함한 반복 사건 분석이 필요하다.
V. 임종 전 가설과 종점 정렬 경향
관찰된 역시간 경향
점 크기는 해당 주에 포함된 환자 수를 나타내고 세로선은 환자 군집 95% 구간이다.
사망 정렬 주별 음성률은 대체로 낮은 수준에 머물며 마지막 1–4주에
지속적인 상승을 보이지 않는다. 독립 검열 경향은 변동이 크고 마지막
주에 44.93%까지 상승하지만, 일부 이전 주에는 포함 환자가 적다.
4주 비교에서 통합값과 짝지은 값은 서로 다르다
막대는 통합 환자 가중 분율이다. 아래의 짝지은 평균 차이는 두 창에 모두 존재한 환자만 사용한다.
사망 정렬 짝지은 비교
−8.53%p
짝지은 환자 28명
95% 구간 −14.08부터 −2.69%p
독립 검열 짝지은 비교
−2.05%p
짝지은 환자 15명
95% 구간 −15.07부터 +9.53%p
독립 검열군의 통합 환자 가중 분율은 30.34%에서 36.77%로 상승하지만,
환자 내 짝지은 평균 변화는 −2.05%p이다. 이는 산술적 모순이 아니다.
통합값과 짝지은 값은 서로 다른 환자 구성과 가중 방식을 사용하며,
두 창에 모두 존재한 독립 검열 환자는 15명뿐이다.
사망 전 4주 또는 8주의 안정적인 경계는 발견되지 않았다
양수는 말기 음성률 상승, 음수는 말기 음성률 하락을 의미한다.
코호트
선택 경계
차이
변화를 지지하는 ΔBIC
부트스트랩 지지율
근거
사망
3주
−4.74%p
−1.215
23.60%
지지되지 않음
독립 검열
2주
+10.59%p
−2.115
37.80%
지지되지 않음
사망 경계는 부트스트랩 표본에서 3–10주 사이로 넓게 변하고 변화량
구간도 음수와 양수를 모두 포함한다. 따라서 현재 자료에서는 사망 전
4주 또는 8주에 재현 가능한 전환점을 확인할 수 없다.
조정 선형 경향
독립 검열에 4주 가까워짐
OR 1.426
95% 구간 1.086–1.873
사망에 4주 가까워짐
OR 0.904
95% 구간 0.695–1.177
사망 대 검열 경향 비
OR 0.634
95% 구간 0.434–0.927
채취 강도
OR 1.826
95% 구간 1.212–2.752
사망 경사 하나보다 상호작용이 더 중요하다. 종점이 가까워질 때
음성률이 변하는 방식이 두 코호트에서 다르다. 채취 강도 역시 음성
결과와 연관되어 있어 관찰 과정이 중립적이지 않을 가능성이 있다.
예측력과 단순성의 충돌: GAMM이 선택된 이유
혼합효과 로지스틱
고정효과 14개
AICc 1026.204
BIC 1099.624
환자 CV 로그손실 0.645675
단순성과 정보기준에서 우세
⇄
일반화 가법 혼합모델
고정효과 24개
AICc 1031.765
BIC 1153.620
환자 CV 로그손실 0.622606
설정된 예측 개선 기준으로 선택
GAMM은 환자 단위 교차검증 로그손실을 0.0231, 상대적으로 약 3.6%
낮춘다. AICc와 BIC는 단순 모델을 선호하지만 환자 외 예측은 비선형
곡선을 선호한다. 따라서 GAMM은 생물학적 기전을 입증한 모델이 아니라
선호되는 예측 모델로 기술하는 편이 적절하다.
조정 베이지안 경향
음영은 라플라스 근사 사후분포의 95% 신용구간이다.
사후 대비
평균 변화
95% 신용구간
증가 확률
사망 말기−이전
−2.24%p
−8.19부터 +3.58%p
22.85%
독립 검열 말기−이전
+9.19%p
+1.74부터 +16.49%p
99.33%
사망 변화−검열 변화
−11.44%p
−20.99부터 −1.76%p
P(사망 변화 > 검열 변화) = 1.10%
독립 검열 짝지은 결과와 조정 베이지안 결과가 다른 것은 서로 다른
질문에 답하기 때문이다. 짝지은 분석은 두 창에 모두 존재한 환자
15명만 사용한다. 조정 모델은 환자 60명의 배양 1,019건을 사용하고,
공변량 보정과 계층적 수축을 적용한다. 여러 모델에서 공통으로 남는
결론은 사망 경향에 독립 검열과 같은 말기 음성률 상승이 없다는 점이다.
말기 결과가 입증하지 않는 것
지지되는 표현
실제 채취된 배양에서는 사망에 가까워질수록 음성률이 상승한다는 근거가 없다.
지지되지 않는 표현
말기 질환이 지속 양성을 유발하거나 생물학적 음전을 막는다.
남아 있는 관찰 편향
종점 근처에서 검체 확보, 채취 강도, 검체 부위 구성이 변할 수 있다.
VI. P / N1 / N2 / C / 사망 경쟁위험 모델
코호트와 최초 사건
199
포함 에피소드
77
고유 환자
54
확인 C 우선
83
사망 우선
62
우측 검열
C 우선
27.14%
사망 우선
41.71%
우측 검열
31.16%
이는 관찰된 에피소드 구성이다. 에피소드의 약 3분의 1이 검열되므로
장기 흡수확률과 동일하지 않다.
최초 사건 상태 구조
P
⇄
N1
⇄
N2
확인된 국소 C
흡수 최초 사건
사망
경쟁 흡수 사건
C 이후 재양성은 이 최초 사건 모델 밖에 있다. 독립 비사망 종점은
우측 검열로 처리된다.
전이 강도는 불규칙한 관찰 시간을 보존한다
배양 1회당 확률이 아니라 출발 상태의 100 노출일당 전이율이다.
현재 상태
전진율
초기화율
사망률
해석
P
P → N1: 0.8612
P 유지는 계속되는 노출 시간
0.3224
지속 양성에서 벗어나는 속도가 느리다.
N1
N1 → N2: 2.7965
N1 → P: 2.4751
0.2893
진행과 초기화가 비슷하게 경쟁한다.
N2
N2 → C: 2.5776
N2 → P: 1.1933
0.1909
다음 이탈은 확인 C가 가장 우세하다.
현재 상태에 따라 장기 C-선행-사망 확률이 크게 달라진다
음성 근거가 누적될수록 장기 최초 사건의 균형이 C 방향으로 이동한다.
출발 상태
C가 사망보다 먼저
95% 구간
사망이 C보다 먼저
P
42.07%
30.29%–52.89%
57.93%
N1
57.81%
45.77%–68.52%
42.19%
N2
77.73%
66.65%–85.57%
22.27%
P에서 시작한 고정 시점 확률
고정 시점 추정은 아직 P/N1/N2에 남아 있는 미해결 분율을 보존한다.
P에서 시작
확인 C
사망
P/N1/N2 잔류
30일
1.32%
9.05%
89.63%
60일
5.51%
16.83%
77.67%
90일
10.59%
23.41%
66.00%
180일
23.22%
37.52%
39.26%
365일
35.67%
51.01%
13.32%
P에서 C가 사망보다 먼저 일어날 장기 확률 42.07%와 90일 C 확률
10.59%는 서로 다른 질문이다. 장기 확률은 흡수될 때까지 모델을
계속 진행하지만, 90일 결과에서는 66.00%가 아직 미해결 상태이다.
모델의 경계
보존되는 요소
불규칙한 위험 시간
경쟁 최초 사건
환자 군집 부트스트랩 불확실성
명시적인 동일 날짜 처리 규칙
직접 모델링하지 않는 요소
정확한 구간 검열 전이 시점
체류 시간에 따라 변하는 전이율
C 이후 반복 사건
시간에 따라 변하는 감시 강도
VII. 생물학적 HMM과 전환 HMM
생물학적 HMM: 단계 경사는 일관되지만 초기 상태에 매우 민감하다
설계에 맞춘 1차 프로파일
BIC 2206.435
양성 등록 후 기준 잠재 상태를 Colonized로 고정.
추정 초기 상태
BIC 2187.042
1차 모델보다 BIC가 19.392점 낮다.
정상분포 초기 상태
BIC 2179.351
BIC 선호 프로파일; 27.084점 개선.
모든 프로파일은 수렴하고 P에서 C까지 단조 감소를 유지하지만, 기준
잠재 상태 가정이 적합도와 모수, 단계별 사후확률을 의미 있게 바꾼다.
따라서 생물학적 HMM은 탐색적으로 유지해야 한다.
전환 HMM 차수 비교
BIC와 CV 로그손실은 낮을수록 좋다. 5체제 모델은 수렴하지 않았다.
모델
수렴
AICc
BIC
CV 로그손실
최소 점유율
최소 분리도
4상태 MM / 1체제
예
2254.700
2288.967
0.506774
100.00%
NE
2체제
예
2164.659
2250.204
0.486243
31.64%
34.13%
3체제
예
2173.920
2321.938
0.483468
27.59%
12.57%
4체제
예
2193.655
2415.218
0.482243
13.15%
10.74%
5체제
아니오
2224.257
2530.284
0.483921
5.15%
12.69%
4체제는 2체제보다 CV 로그손실을 0.0040, 약 0.82%만 개선하지만
BIC는 약 165점 악화되고 전이 프로파일 분리도도 낮아진다. 따라서
2체제가 더 방어 가능한 절충안이다.
두 숨은 체제의 형태
체제 2의 전진 및 C 유지 영역이 체제 1보다 현저하게 크다.
특징
체제 1
체제 2
P → N1
7.55%
54.34%
N1 → N2
28.51%
69.77%
N2 → C
42.07%
79.30%
C → C 유지
16.46%
61.45%
관찰 점유율
68.36%
31.64%
정상확률
10.56%
89.44%
체제 1은 낮은 진행과 높은 P 복귀 패턴이고, 체제 2는 높은 진행과
더 나은 C 유지 패턴이다. 이는 전이 역학을 설명하는 명칭이며 고정된
환자 표현형으로 바꾸어서는 안 된다.
단순 2상태 모델이 참고 분석으로만 남아야 하는 이유
전이 동질성
p = 2.536 × 10−21
하나의 통합 과정은 지지되지 않는다.
기억 없음
p = 3.657 × 10−11
현재의 이분 결과만으로는 이력이 부족하다.
마르코프 예측 점검
4개 중 2개 경고
절대 적합성이 제한적이다.
2상태 HMM 점검
4개 중 4개 경고
상대적 개선으로 충분성을 회복하지 못한다.
2상태 HMM은 2상태 마르코프 모델보다 AICc를 70.642점, BIC를
58.932점 개선하고 CV 로그손실도 2.53% 낮춘다. 그러나 두 모델 모두
중요한 구조 또는 예측 점검에 실패한다. 따라서 “두 모델 모두
충분하지 않음”이라는 결론이 적절하다.
VIII. 구체적 시퀀스 예와 신뢰도 검토
구체적 예: 환자 1001, CRE 대변
관찰된 배양은 모두 양성이었고 시퀀스는 전체 기간 동안 P에 머물렀다.
날짜
배양
간격
운영 상태
HMM P(colonized)
2024-02-20
양성
기준
P → P
99.14%
2024-02-27
양성
7일
P → P
99.52%
2024-03-04
양성
6일
P → P
99.61%
2024-03-07
양성
3일
P → P
99.60%
2024-03-14
양성
7일
P → P
99.47%
2024-03-21
양성
7일
P → P
98.96%
2024-03-26
양성
5일
P → P
97.00%
이 시퀀스는 관찰 상태와 잠재 상태의 차이를 보여준다. 모든 배양이
양성이므로 운영 상태는 P로 유지된다. HMM 사후확률은 적합된 잠재
과정 때문에 조금씩 변하지만 매우 높은 상태이며 음전을 뜻하지 않는다.
구체적으로 수정하거나 검토해야 할 항목
우선도
문제
중요한 이유
권장 수정
치명적
서로 다른 출처 단계에 50건, 19건, 30건의 차이가 남아 있다.
분석 실행 성공만으로 원자료 완전성을 입증할 수 없다.
관찰 단위 조정 원장을 만든다.
치명적
시퀀스 준비에 오류 수준 문제 8건이 있다.
모든 4상태 및 후속 모델이 시퀀스 재구성에 의존한다.
영향 시퀀스, 원본 문장, 규칙, 포함 여부를 표시한다.
높음
혼합모델 주별 구간이 0%–100%로 표시된다.
정보성이 없으며 예측 분산 대상이 잘못 계산되었을 수 있다.
모집단 신뢰구간, 신규 환자 예측구간, 조건부 구간을 분리한다.
높음
혈액 대 대변 OR가 약 5,164만이고 SE가 2,308.8이다.
혈액 관찰 7건으로 희소자료 또는 분리 불안정성이 발생한다.
NE로 억제하거나 수준을 결합하거나 명시적 벌점 모델을 사용한다.
중간
독립 검열 짝지은 분석에 환자 15명만 포함된다.
말기 추정치가 부정확하고 코호트 구성에 민감하다.
모든 추정치 옆에 분모를 유지한다.
중간
전환 HMM 차수에 대한 반복 환자 단위 CV가 실행되지 않았다.
한 번의 폴드 배정만으로 차수 안정성을 확정할 수 없다.
여러 시드에서 반복하고 차수별 선택 빈도를 요약한다.
중간
일부 희소 층에 100% 전이가 표시된다.
시각적 강조가 확실성으로 오해될 수 있다.
분자, 분모, 추정 가능성 경고를 함께 표시한다.
형식
1022|CRE|Stool 같은 식별자가 Markdown 표를 깨뜨린다.
세로 막대가 열 구분자로 해석된다.
1022\|CRE\|Stool 또는 1022 · CRE · Stool을 사용한다.
근거 강도 지도
직접적인 지지가 비교적 강함
4상태 전이 수
종점 연결과 출처
재양성 분모
관찰된 주별 말기 음성률
주의를 동반한 지지
조정 혼합모델과 GAMM 경향
2체제 전환 HMM
환자 군집 부트스트랩 구간
기술적이며 추적에 의존
C 이후 재양성
희소 병원체–부위 비교
말기 짝지은 창 추정
탐색적 또는 지지되지 않음
생물학적 HMM 모수
말기 변화점
경쟁위험 흡수확률
단순 2상태 모델의 충분성
IX. 최종 해석과 다음 우선순위
현재 합리적으로 결론 내릴 수 있는 내용
결론
현재 평가
첫 번째 적격 음성이 가장 큰 운영적 병목이다.
기술적으로 지지됨
N1과 N2는 서로 다른 근거 이력을 가지므로 분리해야 한다.
기술적으로 지지됨
세 번째 음성은 의미 있는 확인 단계이다.
기술적으로 지지됨
확인된 국소 음전 이후 양성이 다시 나타날 수 있다.
감시가 계속된 조건에서 지지됨
사망 전에 배양 음성률이 상승한다.
지지되지 않음
2개의 숨은 전이 체제는 단일 체제 MM보다 예측을 개선한다.
주의를 동반해 지지됨
숨은 체제는 고정된 생물학적 환자 유형이다.
입증되지 않음
국소 C는 생물학적 완전 제거 또는 격리 해제를 입증한다.
입증되지 않음
후속 작업의 권장 순서
자료 감사를 종결한다.
원고 산술 차이, 파서 차이, 준비 단계 30건 감소, 시퀀스 오류
8건을 모두 해결한다.
불확실성 출력을 수정한다.
혼합모델의 0%–100% 구간을 바로잡고 혈액 대 대변과 같은 불안정
희소 계수를 억제한다.
환자 단위 검증을 반복한다.
여러 폴드와 시드에서 교차검증을 반복하고 2체제, 3체제, 4체제의
선택 빈도를 요약한다.
관찰 과정을 모델링한다.
사망 또는 독립 검열에 가까워질수록 검체 채취 여부, 부위, 채취
강도가 변하는지 평가한다.
반복 사건 분석을 개발한다.
위험 시간, 검열, 환자 군집을 포함하여 최초 및 반복 재양성을 추정한다.
다상태 모델을 확장한다.
P, N1, N2 체류 시간에 따라 전이율이 달라질 수 있는 준마르코프
또는 구간 검열 모델을 평가한다.
외부 검증을 시행한다.
생물학적 HMM 방출확률, 숨은 체제, C-선행-사망 확률은 다른
코호트에서 재현되기 전까지 탐색적으로 유지한다.
논문에 사용할 수 있는 실질적 결론
본 장기요양 감시 코호트에서 확인된 국소 음전에 이르는 가장 큰
운영적 장벽은 첫 번째 적격 음성 배양을 얻는 단계였다. 음성 근거가
누적된 이후에는 전진 가능성이 높아졌으며, 이는 N1과 N2를 서로 다른
근거 이력 상태로 유지하고 세 번째 적격 음성을 의미 있는 확인 단계로
보존하는 해석을 지지한다. 감시가 계속된 조건에서 확인된 국소 음전은
항상 지속되지 않았으나, 재양성 추정치는 분석 단위와 음전 후 추적
가능성에 크게 의존하였다. 배양 결과로 정의된 종점을 독립 검열
종점과 분리한 이후, 사망 정렬 배양에서는 음성률의 말기 상승이
지지되지 않았다. 4상태 운영 모델은 1차 해석 모델로 유지하는 편이
타당하다. 2체제 전환 HMM은 2차 예측 모델로 사용할 수 있으나,
생물학적 HMM 모수, 변화점 추정치, 경쟁위험 흡수확률은 명시적으로
탐색 결과로 유지해야 하며 격리 해제 또는 치료 기준으로 사용해서는 안 된다.
Written on July 30, 2026
Assessment of the latest nGeneMDRO analysis (Written July 31, 2026)
Overall assessment:
The analytical engine, corrected outcome provenance, patient-specific pre-death windows,
and the prespecified four-week and eight-week tests all operated successfully.
The present data do not support a sudden increase in negative cultures before death.
The remaining work concerns report completeness, source-row auditing, unstable uncertainty output,
and final regression validation rather than reconstruction of the analytical concept.
I. One-page research map
SOURCE DATA
98 patients
2,703 parsed cultures
RawDataset.zip
→
OUTCOME PROVENANCE
44 / 37 / 17
Death / independent censoring / culture-defined
98 of 98 linked
→
CORE CLEARANCE MODEL
234 sequences
P → N1 → N2 → C
88 reached C
→
PRE-DEATH WINDOWS
34 profiles
Final 4 vs previous 4
Final 8 vs previous 8
→
COMPETING RISK
199 episodes
54 C-first / 83 Death-first
77 patients
→
REPORT AND VALIDATION
29 / 34 sections
8 sequence errors
38 outcome warnings
ANALYTICAL ENGINE
Operating successfully
All 11 pre-death and terminal analysis families completed.
PRIMARY HYPOTHESIS
Directly tested
Equal-length four-week and eight-week windows were compared.
SCIENTIFIC RESULT
Terminal increase not supported
The final-four estimate was lower and the final-eight estimate remained uncertain.
PUBLICATION READINESS
Not yet complete
Export, source-row audit, uncertainty output, and optional diagnostics remain.
Domain
Current status
Key evidence
Reading
Data pipeline
Completed with review items
3 of 3 families completed
The pipeline ran, but eight sequence-preparation errors remain.
Core clearance models
Completed
6 of 6 families completed
The Four-State model remains the primary protocol-aligned model.
Pre-death and terminal models
Completed
11 of 11 families completed
The requested four-week and eight-week hypotheses were directly tested.
Competing-risk models
Completed
4 of 4 families completed
Confirmed C and Death were treated as competing first-event outcomes.
Optional diagnostics
Not run
0 of 1 family completed
The final validation package remains incomplete.
II. How patients move through the pre-death analysis
98 Culture patients linked
→
81 Primary endpoint records
→
44 Verified deaths
→
34 Eligible pre-death profiles
→
19 Complete final-four windows
→
16 Four-week paired test
→
10 Eight-week paired test
Stage
Patients
Why the denominator changes
Interpretive role
All linked culture patients
98
Every culture patient had one resolved outcome-provenance record.
Outcome-linkage denominator
Primary endpoint records
81
Seventeen culture-defined endpoints were retained for audit but excluded from the primary comparator.
Death plus independent censoring
Verified deaths
44
Only death-linked patients can contribute to death-relative windows.
Death cohort
Eligible pre-death profiles
34
Ten death-linked patients lacked an eligible culture panel on or before death.
Descriptive patient-window cohort
Complete final-four windows
19
All four scheduled positions D−1 through D−4 had to be observed.
Complete terminal-window coverage
Four-week paired contrast
16
Both D−1–4 and D−5–8 had to be complete.
Confirmatory four-week test
Eight-week paired contrast
10
All component weeks D−1 through D−16 had to be represented.
Confirmatory eight-week test
A smaller paired denominator is not automatically evidence of data loss.
It reflects the requirement that the same patient be observed in both equal-length windows.
Patients with incomplete windows remain visible descriptively but do not enter the confirmatory comparison.
III. The exact pre-death question
D−13–16
Earlier reference block
D−9–12
Later reference block
D−5–8
Immediately previous four weeks
D−1–4
Final four scheduled weeks
Death
Four-week surge hypothesis
D−1–4 minus D−5–8 should be greater than zero.
Eight-week elevation hypothesis
D−1–8 minus D−9–16 should be greater than zero.
Window
Scheduled-week positions
Question answered
Required completeness
Final four weeks
D−1 through D−4
Was there an immediate terminal rise?
Four observed scheduled positions
Previous four weeks
D−5 through D−8
What was the immediately preceding level?
Four observed scheduled positions
Final eight weeks
D−1 through D−8
Was the broader terminal period elevated?
Eight observed scheduled positions
Previous eight weeks
D−9 through D−16
What was the equal-length earlier reference?
Eight observed scheduled positions
Enrollment anchor
E1 through E4
How did terminal windows compare with early follow-up?
Verified enrollment date or documented first-culture fallback
The weekly observations are noisy rather than smoothly progressive.
The death-aligned curve does not show a consistent late rise.
The final four death-aligned weeks remain around 11%–17% on the patient-weighted scale,
whereas the censoring-aligned curve rises more clearly in its final week.
IV. What the pre-death results show
Analysis
Estimate
Interval
Directional evidence
Visual reading
Conclusion
Final 4 vs previous 4
−4.78 pp
−12.60 to +3.81 pp
One-sided P = 0.8671
Decrease direction
No terminal four-week surge
Final 8 vs previous 8
+3.85 pp
−11.98 to +20.94 pp
One-sided P = 0.3355
Uncertain increase direction
Elevation not confirmed
Weeks 1–4 vs weeks 5–12
−8.53 pp
−14.08 to −2.69 pp
Bootstrap interval below zero
Supported decrease
Negative fraction was lower nearer death
Bayesian death terminal vs earlier
−2.24 pp
−8.19 to +3.58 pp
P(increase) = 22.85%
Decrease favored
Posterior evidence does not favor an increase
Death change minus censoring change
−11.44 pp
−20.99 to −1.76 pp
P(death change > censoring change) = 1.10%
Death-specific increase rejected
The death-aligned change was smaller than the censoring-aligned change
FINAL FOUR WEEKS
−4.78 pp
The point estimate moved in the opposite direction from the hypothesis.
FINAL EIGHT WEEKS
+3.85 pp
The interval crossed zero widely, so the apparent increase was not supported.
BAYESIAN PROBABILITY
22.85%
Only about one posterior draw in four supported a death-terminal increase.
CHANGE POINT
Not supported
The selected death boundary had ΔBIC −1.215 and only 23.60% bootstrap support.
Central scientific conclusion:
The current dataset does not support the hypothesis that negative cultures suddenly become more frequent
during the final four or final eight scheduled weeks before death.
The most direct final-four estimate is lower, the final-eight estimate is uncertain,
the broader paired death-window comparison is significantly lower,
the death change point is unsupported,
and the Bayesian death-specific comparison also argues against a terminal increase.
What this result means
The prespecified terminal-surge hypothesis was tested directly.
The available evidence does not support the hypothesized increase.
A negative or unsupported result remains scientifically useful.
The paired denominator must remain visible in every report.
What this result does not mean
It does not prove that terminal illness has no microbiological effects.
It does not treat missing specimens as negative cultures.
It does not establish a causal mechanism.
It does not replace source-row quality control.
V. Why the outcome correction matters
EARLIER CLASSIFICATION
54
All non-death endpoints labeled as censoring
→
CORRECTED CLASSIFICATION
37
Independent censoring
17
Culture-defined endpoints
Measure
Earlier result
Corrected result
Change
Methodological interpretation
Independent censoring patients
54
37
−17
Culture-defined endpoints were removed from the neutral comparator.
Culture-defined endpoints
Not separated
17
Explicitly identified
These remain visible for audit and sensitivity analysis.
Primary-analysis endpoints
98 implicitly used
81
−17
The primary endpoint cohort now contains only 44 deaths and 37 independent censoring records.
Competing-risk episodes
234
199
−35
Culture-derived terminal dates no longer enter the fitted primary episode model.
Competing-risk patients
91
77
−14
The revised cohort follows the corrected endpoint-provenance rule.
Confirmed-C-first episodes
88
54
−34
Culture-defined endpoint episodes are no longer treated as primary-model follow-up.
Death-first episodes
83
83
0
The death count remained stable, supporting the internal consistency of the correction.
The smaller corrected competing-risk cohort is expected rather than accidental.
The excluded records were not lost; they remain visible as culture-defined endpoint provenance
but are no longer used as if they were independent censoring.
VI. What the core MDRO models add
The Four-State model shows where clearance progression is difficult
P
Positive status
16.78%
→
Main bottleneck
N1
First negative
55.64%
→
Forward progression
N2
Two negatives
73.10%
→
Third-negative confirmation
C
Confirmed clearance
N1 → P reset: 44.00%
N2 → P reset: 26.90%
C → P recurrence transition: 42.03%
IMMEDIATE CLEAN RUN
6.82%
One uninterrupted P → N1 → N2 → C path
EVENTUALLY REACHED C
37.61%
Later attempts and resets are included
MEDIAN CONFIRMATION
48.5 days
Median time to third-negative confirmation
The biological HMM shows a strong stage gradient but remains assumption-sensitive
Operational stage
Mean P(colonized)
Directional meaning
Important limitation
P
92.53%
Strong latent-colonization signal
The value depends on the initial hidden-state profile.
N1
25.50%
Large reduction after one qualifying negative
One negative remains operationally fragile.
N2
2.96%
Two negatives provide strong evidence
N2 is not itself confirmed clearance.
C
0.94%
Very low modeled colonization probability
The model remains exploratory because initial-state sensitivity is high.
The competing-risk model separates confirmed clearance, death, and unresolved follow-up
Starting state
Confirmed C by 90 days
Death by 90 days
Still P/N1/N2
Reading
P
10.59%
23.41%
66.00%
Most episodes remain unresolved at 90 days.
N1
34.13%
18.11%
47.76%
One negative improves the clearance trajectory but leaves substantial uncertainty.
N2
65.25%
10.04%
24.71%
Two negatives strongly shift the first-event probability toward confirmed C.
Eight sequence-preparation errors require a source-row audit
The analysis pipeline completed, but the manifest still labels Sequence Preparation as
Not Supported. Each affected row should have a documented retain, merge,
exclude, or conflict-resolution decision.
No outcome row was classified as a parser error, but 38 warnings remain.
The report does not expose the exact warning list in this export,
so the warning panel should be reviewed directly rather than inferred.
Mixed-model prediction intervals remain too broad
Repeated 0%–100% intervals do not provide useful inferential information.
Such intervals should be recalculated, suppressed as NE, or explicitly identified as non-informative.
The Blood site coefficient is structurally unstable
Only seven Blood observations are present.
The resulting coefficient and standard error are too unstable for ordinary standalone interpretation.
Pooling, shrinkage, or suppression is preferable.
The complete export still omits the five new patient-window sections
The schema label has advanced to v4 and the manifest contains the new analyses,
but the section count remains 29 rather than approximately 34.
The analytical engine and the export route are therefore not yet fully synchronized.
VIII. Recommended completion path
STEP 1
Audit source issues
Review 8 sequence errors and 38 outcome warnings.
→
STEP 2
Repair report export
Confirm all five patient-window sections.
→
STEP 3
Verify window displays
Check D−1–16, E1–4, Entire, and patient profiles.
→
STEP 4
Stabilize adjusted models
Resolve 0%–100% intervals and sparse Blood estimates.
→
STEP 5
Run diagnostics and tests
Run optional diagnostics, XCTest, and deterministic regression tests.
→
STEP 6
Generate the final archive
Re-run the entire pipeline and save a current v4 report.
Priority
Action
Completion criterion
Why it matters
1
Audit sequence and outcome notices
Every affected source row has a documented disposition.
Prevents silent provenance loss.
2
Repair complete-report composition
Approximately 34 sections and all five patient-window keys appear.
Makes the exported result match the completed analytical manifest.
3
Verify enrollment and individual-patient outputs
D−1–4, D−5–8, D−9–12, D−13–16, E1–4, and Entire are visible.
Confirms the full requested patient-level comparison.
4
Stabilize sparse adjusted-model terms
Non-informative intervals are suppressed or corrected; Blood is pooled or regularized.
Prevents unstable coefficients from appearing definitive.
5
Run optional diagnostics and regression tests
Optional diagnostics complete and all ISO-week tests pass.
Validates the implementation rather than only the numerical output.
6
Re-run and archive
No stale result, current schema v4, complete sections, reviewed warnings.
Creates a defensible final research artifact.
Analytical design
Successfully implemented
Four-week and eight-week tests
Successfully completed
Terminal negative-culture surge
Not supported
Final publication package
Further validation required
Final judgment:
The most important analytical objective has been achieved.
The pre-death hypothesis is now tested with the intended scheduled-week structure,
endpoint provenance is corrected,
and the principal analyses consistently indicate no sudden increase in negative cultures before death.
The remaining tasks are quality-control and reporting tasks rather than reconstruction of the core analysis.
최신 nGeneMDRO 분석 진행 상태 평가
전체 판정:
분석 엔진, 교정된 outcome provenance, 환자별 임종 전 window,
사전에 정한 4주 및 8주 검정은 모두 정상적으로 작동하였습니다.
현재 자료는 임종 전에 음성 culture가 갑자기 증가한다는 가설을 지지하지 않습니다.
남은 작업은 분석 개념의 재구성이 아니라 report 완결성, source-row audit,
불안정한 uncertainty 출력, 최종 regression validation입니다.
I. 한눈에 보는 연구 구조
원자료
환자 98명
Culture 2,703건
RawDataset.zip
→
OUTCOME PROVENANCE
44 / 37 / 17
사망 / 독립적 censoring / culture-defined
98명 전원 linkage
→
핵심 CLEARANCE MODEL
Sequence 234개
P → N1 → N2 → C
88개가 C 도달
→
임종 전 WINDOW
Profile 34명
마지막 4주 대 직전 4주
마지막 8주 대 직전 8주
→
COMPETING RISK
Episode 199개
C-first 54 / Death-first 83
환자 77명
→
REPORT와 검증
29 / 34 section
Sequence error 8개
Outcome warning 38개
분석 엔진
정상 작동
임종 전 및 terminal 분석 11개가 모두 완료되었습니다.
주가설
직접 검정 완료
같은 길이의 4주 및 8주 구간이 비교되었습니다.
연구 결과
임종 전 증가 지지되지 않음
마지막 4주는 더 낮았고, 마지막 8주는 불확실했습니다.
논문 준비 상태
아직 미완료
Export, source-row audit, uncertainty 출력, 선택적 진단이 남아 있습니다.
모든 culture 환자에게 하나의 outcome-provenance record가 연결되었습니다.
Outcome linkage 분모
Primary endpoint
81명
Culture-defined endpoint 17명은 audit에는 남기되 주 비교군에서는 제외했습니다.
사망 + 독립적 censoring
확인된 사망
44명
사망 환자만 death-relative window에 들어갑니다.
사망 cohort
임종 전 profile
34명
10명은 사망일 이전에 적격 culture panel이 없었습니다.
기술적 patient-window cohort
마지막 4주 완전관찰
19명
D−1부터 D−4까지 네 scheduled position이 모두 필요합니다.
완전한 terminal-window coverage
4주 paired contrast
16명
D−1–4와 D−5–8이 모두 완전해야 합니다.
Confirmatory 4주 검정
8주 paired contrast
10명
D−1부터 D−16까지 필요한 모든 구간이 있어야 합니다.
Confirmatory 8주 검정
Paired 환자 수가 작아진 것이 곧 자료 손실을 뜻하지는 않습니다.
같은 환자가 target과 reference 구간에 모두 관찰되어야 하기 때문입니다.
불완전한 환자는 개인 profile에는 남지만 confirmatory comparison에는 들어가지 않습니다.
III. 실제로 검정한 임종 전 가설
D−13–16
더 이른 reference
D−9–12
후반 reference
D−5–8
직전 4주
D−1–4
마지막 4 scheduled week
사망
4주 급증 가설
D−1–4에서 D−5–8을 뺀 값이 0보다 커야 합니다.
8주 상승 가설
D−1–8에서 D−9–16을 뺀 값이 0보다 커야 합니다.
구간
Scheduled-week 위치
답하는 질문
필요한 completeness
마지막 4주
D−1부터 D−4
임종 직전에 즉각적인 상승이 있었는가?
네 개 scheduled position
직전 4주
D−5부터 D−8
바로 앞선 시기의 기준값은 어떠했는가?
네 개 scheduled position
마지막 8주
D−1부터 D−8
더 넓은 terminal 기간이 상승했는가?
여덟 개 scheduled position
직전 8주
D−9부터 D−16
길이가 같은 earlier reference는 어떠했는가?
여덟 개 scheduled position
Enrollment anchor
E1부터 E4
임종 전 window가 초기 follow-up과 어떻게 달랐는가?
확인된 enrollment date 또는 명시된 first-culture fallback
주별 관찰값은 부드럽게 상승하는 곡선이 아니라 상당히 불규칙합니다.
사망군에서는 임종 직전의 일관된 상승이 보이지 않습니다.
마지막 4주의 patient-weighted 값은 대체로 11%–17% 정도이며,
censoring군은 마지막 주에 상대적으로 더 높은 값을 보입니다.
IV. 임종 전 분석 결과의 의미
분석
추정치
불확실성 구간
방향성 근거
시각적 판정
결론
마지막 4주 대 직전 4주
−4.78%p
−12.60부터 +3.81%p
단측 P = 0.8671
감소 방향
마지막 4주 급증 없음
마지막 8주 대 직전 8주
+3.85%p
−11.98부터 +20.94%p
단측 P = 0.3355
불확실한 증가 방향
상승 확인되지 않음
1–4주 대 5–12주
−8.53%p
−14.08부터 −2.69%p
Bootstrap interval이 0 미만
감소 지지
임종에 가까울수록 음성 비율이 더 낮음
Bayesian 사망 terminal 대 earlier
−2.24%p
−8.19부터 +3.58%p
P(상승) = 22.85%
감소 방향 선호
Posterior evidence가 증가를 선호하지 않음
사망 변화량 − censoring 변화량
−11.44%p
−20.99부터 −1.76%p
P(사망 변화가 더 큼) = 1.10%
사망 특이 증가 배제
사망군 변화가 censoring군보다 작음
마지막 4주
−4.78%p
점추정치는 가설과 반대 방향으로 움직였습니다.
마지막 8주
+3.85%p
구간이 0을 넓게 포함하여 증가를 지지하지 못했습니다.
BAYESIAN PROBABILITY
22.85%
Posterior draw 네 개 중 약 한 개만 사망 직전 증가를 지지했습니다.
CHANGE POINT
지지되지 않음
사망군 boundary는 ΔBIC −1.215, bootstrap support 23.60%였습니다.
핵심 연구 결론:
현재 자료는 임종 전 마지막 4주 또는 마지막 8주에 음성 culture가 갑자기 증가한다는 가설을 지지하지 않습니다.
가장 직접적인 마지막 4주 추정치는 낮았고, 마지막 8주 추정치는 불확실했으며,
더 넓은 사망 window 비교에서도 음성 비율이 유의하게 낮았습니다.
사망 change point도 지지되지 않았고 Bayesian 분석 역시 사망 특이적 증가에 반대되는 방향입니다.
이 결과가 의미하는 것
사전에 정한 terminal-surge 가설이 직접 검정되었습니다.
현재 근거는 가설을 지지하지 않습니다.
지지되지 않은 가설도 충분히 의미 있는 연구 결과입니다.
모든 보고서에 paired denominator가 함께 제시되어야 합니다.
이 결과가 의미하지 않는 것
Terminal illness가 미생물학적 영향을 전혀 주지 않는다는 증명은 아닙니다.
Missing specimen을 음성으로 처리한 결과가 아닙니다.
인과적 기전을 확립한 결과가 아닙니다.
Source-row quality control을 대신하지 않습니다.
V. Outcome 교정이 중요한 이유
이전 분류
54명
모든 비사망 endpoint를 censoring으로 분류
→
교정된 분류
37명
독립적 censoring
17명
Culture-defined endpoint
항목
이전 결과
교정 결과
변화
방법론적 의미
독립적 censoring 환자
54명
37명
−17명
Culture-defined endpoint가 중립적 비교군에서 제외되었습니다.
Culture-defined endpoint
별도 분리 없음
17명
명시적으로 분리
Audit와 sensitivity analysis에는 계속 남습니다.
Primary-analysis endpoint
98명 전체가 사실상 사용됨
81명
−17명
사망 44명과 독립적 censoring 37명만 주 분석에 포함됩니다.
Competing-risk episode
234개
199개
−35개
Culture-derived terminal date가 fitted primary model에 들어가지 않습니다.
Non-informative interval이 억제되고 Blood가 pooling 또는 regularization됩니다.
불안정한 coefficient가 확정적인 결과처럼 보이지 않도록 합니다.
5
Optional diagnostics와 regression test
Diagnostics 완료 및 모든 ISO-week test 통과
수치뿐 아니라 구현 자체를 검증합니다.
6
재실행 및 archive
Stale result 없음, current schema v4, complete section, warning 검토 완료
방어 가능한 최종 연구 artifact를 만듭니다.
분석 설계
정상적으로 구현
4주 및 8주 검정
정상적으로 완료
임종 전 음성 급증
지지되지 않음
최종 논문 package
추가 validation 필요
최종 판정:
가장 중요한 분석 목적은 달성되었습니다.
임종 전 가설은 의도한 scheduled-week 구조로 검정되었고,
endpoint provenance도 교정되었으며,
주요 분석들은 임종 전 음성 culture의 갑작스러운 증가가 없다는 방향으로 일관됩니다.
남은 작업은 핵심 분석의 재구성이 아니라 품질관리와 report 완결성 작업입니다.
Written on July 31, 2026
Is the expanded MM–Bayesian publication framework working correctly? (Written August 3, 2026)
Overall verdict: The project is moving in the correct direction. The Four-State Operational MM is established, the two-hidden-regime extension is statistically supported with caution, patient-level outcomes are completely linked, and the Death-inclusive competing-risk model is functioning. The new discrete Five-State comparison and the new Bayesian predictive-superiority test, however, are not yet present in the current canonical report. They are therefore ready to be tested, but not yet proven on the real dataset.
ESTABLISHED
Four-State MM
234 sequences
P → N1: 16.78%
N2 → C: 73.10%
SUPPORTED WITH CAUTION
Two hidden regimes
ΔAICc: +90.04
ΔBIC: +38.76
CV log-loss gain: 4.05%
COMPLETE
Outcome linkage
98 / 98 patients linked
44 Death endpoints
0 date conflicts
FUNCTIONING
Competing-risk model
199 episodes
77 patients
400 / 400 bootstrap runs
REAL-DATA RESULT PENDING
Discrete Five-State MM
Shared Death versus state-specific Death
AICc · BIC · Bayes factor
Posterior Death intervals
REAL-DATA RESULT PENDING
Bayesian predictive value
MLE versus posterior prediction
Held-out log loss · Brier score
Patient-cluster interval
I. Visual verdict: what is established and what remains pending
The data and cohort infrastructure are sufficiently mature for the new analysis
The current report contains a complete data pipeline, deterministic sequence preparation, full patient-outcome linkage, a functioning Four-State process, hidden-regime comparisons, and Death-inclusive first-event modeling. This means the new questions are not being added to an empty framework. They are being added to an already functioning analytical system.
Observation reconciliation
The count axis is intentionally zoomed. The four totals describe different provenance stages and must not be silently collapsed into one number.
Sequence cohort flow
Four-State and hidden-regime models use increasingly specific eligibility rules. Their denominators should therefore remain visible.
Infrastructure layer
Current result
Why it matters for the new extension
Culture parsing
2,703 explicit observations from 98 patients
Provides the source observations for operational-state reconstruction.
Prepared sequences
321 patient × pathogen × site sequences
Defines the candidate sequence universe.
Four-State cohort
234 sequences from 91 patients
Provides the direct P, N1, N2, and C reference process.
Hidden-regime cohort
198 sequences from 79 patients
Provides a directly comparable MM-versus-HMM cohort.
Outcome linkage
98 of 98 patients linked; 44 Death endpoints
Makes a Death-state extension estimable.
Date validation
No endpoint-date conflicts
Prevents impossible pre-culture or post-endpoint transitions.
The central status distinction must remain visually explicit
Question
Current canonical report
Permitted conclusion
Does the Four-State MM describe the operational rule?
Yes
The Four-State MM is the primary protocol-aligned model.
Do two hidden transition regimes improve the Four-State process?
Supported with caution
Two regimes are a supported complementary model.
Can Death be incorporated into a multi-state process?
Yes
The continuous-time competing-risk analysis is functioning.
Is a discrete Five-State MM better than the shared-Death baseline?
Not yet reported
No superiority claim yet.
Does Bayesian posterior prediction outperform plug-in MLE?
Not yet reported
No Bayesian predictive-superiority claim yet.
II. Four-State MM: the established reference model
The state diagram directly represents the three-negative operational rule
The main scientific message is immediately visible: leaving P is difficult, one negative remains fragile, and the process becomes progressively more favorable after negative evidence accumulates.
The first qualifying negative is the major bottleneck
Key transition probabilities
The denominator changes the recurrence estimate
Result
Numerical value
Correct interpretation
P → N1
16.78%
The next included culture starts a qualifying negative run.
N1 → N2
55.64%
A second qualifying negative follows N1.
N2 → C
73.10%
A third qualifying negative confirms local clearance.
Immediate clean-run product
6.82%
Probability of one uninterrupted P → N1 → N2 → C path.
Eventually reached C
37.61%
Sequences may persist, reset, and make later attempts before reaching C.
Critical reading rule: The 6.82% uninterrupted product and the 37.61% eventual clearance fraction are different estimands. They must never be presented under the same endpoint label.
III. Does a two-hidden-regime HMM explain the Four-State process better?
The answer is currently yes, as a complementary model
Fit, prediction, and complexity balance
Lower and farther left is preferable. Bubble size represents parameter count. The two-regime model provides the most favorable overall balance.
How the two regimes differ
Regime 1 has lower clearance-progression tendency. Regime 2 has substantially higher progression through N1, N2, and C.
Criterion
Four-State MM
Two-regime HMM
Evidence direction
Parameters
6
15
The HMM must justify nine additional parameters.
AICc
2,254.700
2,164.659
ΔAICc = 90.041 for two regimes
BIC
2,288.967
2,250.204
ΔBIC = 38.763 for two regimes
Patient-level CV log loss
0.506774
0.486243
4.05% relative improvement
Minimum occupancy
Not applicable
31.64%
Neither regime is an almost-empty artifact.
Minimum separation
Not applicable
34.13%
The two transition matrices are materially distinct.
Convergence
Yes
Yes
The selected two-regime model completed estimation.
The hidden layer changes transition laws without replacing P, N1, N2, and C
Publication interpretation: The Four-State MM remains primary for direct interpretation. The two-regime HMM is supported as a complementary heterogeneity model because it improves penalized fit and patient-level prediction without replacing the four observed states.
Why the four-regime model is not selected despite the lowest single CV loss
Model
Parameters
BIC
CV log loss
Minimum occupancy
Selection interpretation
Two regimes
15
2,250.204
0.486243
31.64%
Recommended balance
Three regimes
26
2,321.938
0.483468
27.59%
Small prediction gain with substantially higher complexity.
Four regimes
39
2,415.218
0.482243
13.15%
Best single CV value, but inferior parsimony and weaker occupancy.
Five regimes
54
2,530.284
0.483921
5.15%
Did not converge.
IV. Death modeling: what is already known and what the new Five-State test adds
The existing competing-risk model and the proposed Five-State MM answer different questions
CURRENT REAL-DATA RESULT
Continuous-time competing risk
C and Death are competing first-event outcomes. Exposure days are retained, and the model estimates event rates, absorption probabilities, and future horizons.
NEW REAL-DATA RESULT REQUIRED
Discrete Five-State MM
Each next included transition is modeled directly. Death is absorbing, while C may remain C, recur to P, or proceed to Death.
Feature
Current competing-risk analysis
New discrete Five-State test
Time basis
Exposure days under a continuous-time approximation
Per next included transition
Absorbing outcomes
C and Death are competing first events
Only Death is structurally absorbing
Post-C recurrence
Outside the first-event analysis
C → P remains part of the transition graph
Main question
Does C or Death occur first, and when?
Does Death probability differ according to P, N1, N2, or C?
The existing Death results already show a clinically coherent state gradient
Eventual C-before-Death probability
Starting from P: what happens over time?
42.07%
P starts and C occurs before Death
57.81%
N1 starts and C occurs before Death
77.73%
N2 starts and C occurs before Death
This result is useful background for the Five-State extension. It suggests that accumulated negative evidence is associated with a more favorable first-event trajectory. It does not yet prove that four separate discrete Death probabilities are required.
The new Five-State comparison is a nested test, not a contest between unrelated models
A publication decision must pass through five evidence gates
V. Bayesian modeling: how probability helps and how superiority is proven
The Bayesian network should show the full probability flow
Probability helps in four concrete ways
①
Sparse-row stabilization
Small P/N1/N2/C rows are pulled away from unstable zero or one estimates.
②
Credible intervals
Every transition probability is accompanied by a distribution rather than only a point estimate.
③
Posterior prediction
The next state is represented as a probability vector across P, N1, N2, C, and D.
④
Sensitivity analysis
The conclusion can be tested under weaker and stronger prior concentrations.
The existing Bayesian terminal model demonstrates appropriate statistical restraint
Posterior changes and credible intervals
Intervals crossing zero do not support a directional change. The death-aligned terminal change remains uncertain.
Weekly posterior predictions
The death-aligned curve declines slightly toward the endpoint, while the censoring-aligned curve rises.
Bayesian terminal result
Posterior mean
95% credible interval
Posterior probability
Death terminal minus earlier
−2.24 pp
−8.19 to +3.58 pp
P(increase) = 22.85%
Censoring terminal minus earlier
+9.19 pp
+1.74 to +16.49 pp
P(increase) = 99.33%
Death change minus censoring change
−11.44 pp
−20.99 to −1.76 pp
P(death change > censoring change) = 1.10%
This existing Bayesian result is reassuring because it does not mechanically favor the target hypothesis. The same discipline should govern the new Five-State Bayesian analysis.
Bayesian superiority must be demonstrated on held-out patients
Run the real shared-Death versus Five-State comparison.
○ Required
Run MLE versus Bayesian held-out prediction.
○ Required
Confirm Death-assignment, prior, and repeated-CV sensitivity.
! Must not be claimed yet
“The Five-State MM is superior.”
! Must not be claimed yet
“Bayesian prediction is superior to MLE.”
Final assessment: The difficult conceptual work has been organized correctly. The Four-State MM explains the operational rule, the two-regime HMM adds statistically supported transition heterogeneity, and the competing-risk model demonstrates that Death can be integrated coherently. The remaining task is no longer a vague discussion of whether a more complex model might help. It is a concrete, publication-grade execution of two prespecified tests: shared Death versus state-specific Five-State structure, and plug-in MLE versus Bayesian posterior prediction.
확장된 MM–Bayesian 출판용 프레임워크는 제대로 작동하고 있는가?
종합 판단: 전체 프로젝트는 올바른 방향으로 진행되고 있습니다. Four-State Operational MM은 이미 확립되어 있고, two-hidden-regime 확장은 주의를 전제로 통계적으로 지지되며, 환자 outcome은 완전히 연결되어 있고, Death를 포함한 competing-risk model도 작동하고 있습니다. 다만 새로운 discrete Five-State 직접 비교와 Bayesian predictive-superiority 검정은 현재 canonical report에 아직 결과가 없습니다. 따라서 두 분석은 실제 데이터 검정을 실행할 준비는 되었지만, 아직 우수성이 증명된 상태는 아닙니다.
확립됨
Four-State MM
234개 sequence
P → N1: 16.78%
N2 → C: 73.10%
주의를 전제로 지지됨
Two hidden regimes
ΔAICc: +90.04
ΔBIC: +38.76
CV log-loss 개선: 4.05%
완료
Outcome linkage
98 / 98명 연결
Death endpoint 44명
날짜 충돌 0명
작동 중
Competing-risk model
199개 episode
77명 환자
Bootstrap 400 / 400
실제 데이터 결과 대기
Discrete Five-State MM
Shared Death 대 state-specific Death
AICc · BIC · Bayes factor
Posterior Death interval
실제 데이터 결과 대기
Bayesian predictive value
MLE 대 posterior prediction
Held-out log loss · Brier score
Patient-cluster interval
I. 시각적 핵심 판단: 무엇이 완료되었고 무엇이 남아 있는가?
새 분석을 실행할 데이터와 cohort 기반은 충분히 성숙되어 있습니다
현재 보고서에는 완전한 data pipeline, deterministic sequence preparation, 전체 환자 outcome linkage, Four-State process, hidden-regime comparison, Death-inclusive first-event model이 이미 존재합니다. 따라서 새로운 분석은 기반이 없는 상태에서 추가되는 것이 아니라, 이미 작동하는 분석 체계 위에 추가되는 것입니다.
Observation reconciliation
차이를 잘 보이도록 count axis를 확대했습니다. 네 수치는 서로 다른 provenance stage이므로 하나의 수치로 조용히 통합해서는 안 됩니다.
Sequence cohort flow
Four-State와 hidden-regime model은 점점 더 엄격한 eligibility rule을 사용하므로 denominator가 항상 함께 표시되어야 합니다.
Infrastructure layer
현재 결과
새 확장 분석에서의 의미
Culture parsing
98명에서 2,703개 explicit observation
Operational-state reconstruction의 원자료입니다.
Prepared sequences
321개 patient × pathogen × site sequence
전체 candidate sequence universe를 정의합니다.
Four-State cohort
91명에서 234개 sequence
P, N1, N2, C 직접 기준 모델을 제공합니다.
Hidden-regime cohort
79명에서 198개 sequence
MM과 HMM을 같은 cohort에서 비교할 수 있습니다.
Outcome linkage
98명 중 98명 연결, Death 44명
Death-state 확장을 추정할 수 있게 합니다.
Date validation
Endpoint-date conflict 0명
불가능한 pre-culture 또는 post-endpoint transition을 방지합니다.
현재 결과와 아직 실행 전인 결과를 시각적으로 분리해야 합니다
질문
현재 canonical report
현재 가능한 결론
Four-State MM이 operational rule을 설명하는가?
Yes
Four-State MM을 주 분석 모델로 사용할 수 있습니다.
Two hidden transition regimes가 Four-State process를 개선하는가?
출판 해석: Three-negative rule을 직접 설명하는 주 모델은 Four-State MM으로 유지하는 것이 적절합니다. Two-regime HMM은 네 관찰 상태를 그대로 유지하면서 penalized fit과 patient-level prediction을 개선하므로 transition heterogeneity를 설명하는 보완 모델로 지지됩니다.
Four-regime model의 CV loss가 가장 낮아도 선택하지 않는 것이 적절합니다
모델
Parameters
BIC
CV log loss
Minimum occupancy
선택 해석
Two regimes
15
2,250.204
0.486243
31.64%
권장되는 균형
Three regimes
26
2,321.938
0.483468
27.59%
작은 prediction gain에 비해 complexity가 크게 증가합니다.
Four regimes
39
2,415.218
0.482243
13.15%
단일 CV 값은 가장 낮지만 parsimony와 occupancy가 약합니다.
Five regimes
54
2,530.284
0.483921
5.15%
Convergence 실패
IV. Death modeling: 현재 알고 있는 것과 새로운 Five-State 검정이 추가하는 것
기존 competing-risk model과 새 Five-State MM은 서로 다른 질문에 답합니다
다음 included transition을 직접 모델링합니다. Death만 absorbing이며 C는 C 유지, P recurrence, Death로의 진행을 가질 수 있습니다.
구분
현재 competing-risk analysis
새 discrete Five-State test
시간 기준
Continuous-time approximation의 exposure day
다음 included transition당 확률
Absorbing outcome
C와 Death가 competing first event
Death만 구조적으로 absorbing
Post-C recurrence
First-event analysis 밖에 있음
C → P가 transition graph에 남음
주요 질문
C와 Death 중 무엇이 먼저, 언제 발생하는가?
P, N1, N2, C에 따라 Death 확률이 다른가?
기존 Death 분석은 이미 일관된 state gradient를 보여줍니다
Eventual C-before-Death probability
P에서 시작할 때 시간에 따른 결과
42.07%
P에서 시작할 때 C가 Death보다 먼저 발생
57.81%
N1에서 시작할 때 C가 Death보다 먼저 발생
77.73%
N2에서 시작할 때 C가 Death보다 먼저 발생
이 결과는 Five-State 확장의 중요한 배경입니다. 음성 근거가 축적될수록 더 유리한 first-event trajectory와 연관되어 있음을 보여줍니다. 그러나 네 개의 별도 discrete Death probability가 반드시 필요하다는 것을 아직 증명하지는 않습니다.
새 Five-State 비교는 서로 무관한 모델의 경쟁이 아니라 nested test입니다
\[
H_0:
p_D(P)=p_D(N_1)=p_D(N_2)=p_D(C)
\]
\[
H_1:
p_D(P),\ p_D(N_1),\ p_D(N_2),\ p_D(C)
\text{를 각각 별도로 추정합니다.}
\]
출판 결론은 다섯 개의 evidence gate를 통과해야 합니다
V. Bayesian modeling: 확률이 어떻게 도움이 되며 우월성을 어떻게 증명하는가?
Bayesian network는 전체 probability flow를 보여주어야 합니다
확률적 모델은 네 가지 구체적인 도움을 제공합니다
①
Sparse-row stabilization
작은 P/N1/N2/C row가 불안정한 0 또는 1 추정에서 완화됩니다.
②
Credible interval
모든 transition probability에 point estimate뿐 아니라 distribution이 제공됩니다.
③
Posterior prediction
다음 상태가 하나의 label이 아니라 P, N1, N2, C, D 전체의 probability vector로 표현됩니다.
④
Sensitivity analysis
더 약하거나 강한 prior concentration에서도 결론이 유지되는지 검정할 수 있습니다.
기존 Bayesian terminal model은 적절한 통계적 절제를 보여줍니다
Posterior change와 credible interval
Interval이 0을 통과하면 방향성 변화가 지지되지 않습니다. Death-aligned terminal change는 여전히 불확실합니다.
Weekly posterior prediction
Death-aligned curve는 endpoint에 가까워지며 약간 감소하고, censoring-aligned curve는 증가합니다.
Bayesian terminal result
Posterior mean
95% credible interval
Posterior probability
Death terminal minus earlier
−2.24 pp
−8.19 to +3.58 pp
P(increase) = 22.85%
Censoring terminal minus earlier
+9.19 pp
+1.74 to +16.49 pp
P(increase) = 99.33%
Death change minus censoring change
−11.44 pp
−20.99 to −1.76 pp
P(death change > censoring change) = 1.10%
기존 Bayesian 결과가 target hypothesis를 기계적으로 지지하지 않는다는 점은 오히려 바람직합니다. 새로운 Five-State Bayesian 분석에도 같은 통계적 절제가 적용되어야 합니다.
Continuous-time C-versus-Death competing-risk result
○ 필요
실제 shared-Death versus Five-State comparison 실행
○ 필요
MLE versus Bayesian held-out prediction 실행
○ 필요
Death assignment, prior, repeated-CV sensitivity 확인
! 아직 주장하면 안 됨
“Five-State MM이 더 우수하다.”
! 아직 주장하면 안 됨
“Bayesian prediction이 MLE보다 우수하다.”
최종 평가: 가장 어려운 개념적 구조는 올바르게 정리되어 있습니다. Four-State MM은 operational rule을 설명하고, two-regime HMM은 통계적으로 지지되는 transition heterogeneity를 추가하며, competing-risk model은 Death가 일관된 방식으로 통합될 수 있음을 보여줍니다. 이제 남은 과제는 더 복잡한 모델이 도움이 될 수 있다는 추상적인 논의가 아닙니다. Shared Death versus state-specific Five-State structure와 plug-in MLE versus Bayesian posterior prediction이라는 두 개의 prespecified test를 실제 데이터에서 실행하고 구체적인 결과를 산출하는 일입니다.
Written on August 3, 2026
Is the current nGeneMDRO analysis working, and how should the full result be interpreted? (Written August 4, 2026)
Overall assessment: The analytical pipeline is functioning, all required analysis families have completed, and the latest report reflects a substantially improved model-selection strategy. The strongest defensible conclusion is not that one model has defeated every other model. The current evidence supports a layered model portfolio: the Four-State Operational MM remains the primary interpretable model, the Four-State switching HMM with two hidden regimes is the best-balanced complementary HMM, the continuous-time competing-risk model addresses confirmed clearance versus Death, and the biological HMMs remain exploratory. The direct discrete Five-State Death comparison and the MLE-versus-Bayesian predictive comparison are still absent from the canonical report. The project is therefore functioning well, but the final publication-validation layer remains incomplete.
EXECUTION
Completed
Data pipeline: 3 / 3
Core analyses: 6 / 6
Terminal analyses: 11 / 11
Competing-risk analyses: 4 / 4
DATA AUDIT
Requires review
2,772 → 2,722
2,722 → 2,703
2,703 → 2,673
8 preparation errors
PRIMARY MODEL
Four-State MM
234 sequences
91 patients
2,505 operational observations
Directly interpretable
BEST-BALANCED HMM
Two hidden regimes
ΔAICc: +90.04
ΔBIC: +38.76
CV log-loss gain: 4.05%
CV Brier gain: 3.79%
DEATH ENDPOINTS
Fully linked
98 / 98 patients
44 Death endpoints
37 independent censoring endpoints
0 chronology conflicts
COMPETING RISK
Functioning
199 episodes
77 patients
54 C-first · 83 Death-first
400 / 400 bootstrap runs
DISCRETE FIVE-STATE
Result pending
Shared Death versus
state-specific Death
AICc · BIC · Bayes factor
Posterior Death intervals
BAYESIAN MM / HMM
Utility untested
MLE versus posterior prediction
Held-out log loss
Brier · calibration
Prior sensitivity
I. Direct answer: is the project working correctly?
The analytical execution layer is functioning
Every required data-pipeline, core-analysis, terminal-analysis, and competing-risk family completed. The canonical report contains 29 analytical sections and records a completed required-execution status. This strongly supports that the underlying analytical engine, model execution, result persistence, and report assembly are functioning.
Execution completeness
Completed
Confirms that the requested analytical families finished running.
Evidence quality
Mixed
Some results are supported, some exploratory, and some explicitly not supported.
Report inclusion
29 sections
Confirms documentation coverage, not automatic scientific validity.
Layer
Current status
What the status proves
What it does not prove
Data pipeline
3 / 3 completed
Parsing, preparation, and reconciliation workflows executed.
Every source discrepancy has been resolved.
Core analyses
6 / 6 completed
Four-State and HMM analyses produced results.
Every model is biologically valid.
Terminal analyses
11 / 11 completed
Prespecified and exploratory endpoint analyses ran.
The target terminal hypothesis was supported.
Competing risk
4 / 4 completed
Death-inclusive exposure-time modeling works.
A discrete Five-State MM is superior.
Optional diagnostics
0 / 1 completed
The optional diagnostic remains identifiable as pending.
Model-order stability has been fully established.
The latest model-selection wording is more scientifically defensible
The earlier output emphasized that the two-regime HMM was supported over the Four-State MM for prediction. The latest output now separates the roles more precisely: the Four-State MM remains the primary protocol-aligned model, and the two-regime HMM becomes a supported complementary model for transition prediction and latent transition heterogeneity.
Decision component
Earlier framing
Current framing
Why the change is better
Primary model
HMM appeared to be the overall winner.
Four-State MM remains primary.
Preserves direct interpretation of the three-negative protocol.
HMM role
Supported over the Four-State MM.
Supported complementary model.
Separates prediction and heterogeneity from endpoint definition.
Probability accuracy
CV log-loss improvement emphasized.
CV log loss and Brier improvement both displayed.
Uses two proper scoring rules rather than one.
Universal winner
Potentially ambiguous.
No universal single winner.
Reflects the fact that interpretability and prediction are distinct goals.
“Mixed evidence” is an appropriate result rather than a failure
The report is stronger because negative and uncertain results remain visible. A publication-quality analytical system should distinguish supported, exploratory, inconclusive, and unsupported findings rather than forcing a positive conclusion.
II. Data foundation: strong linkage, but unresolved provenance
The four observation totals represent different stages
Stage
Count
Difference from prior stage
Meaning
Required action
Manuscript stated total
2,772
Reference
Total stated in the manuscript.
Retain as source provenance.
Manuscript pathogen subtotal
2,722
−50
Arithmetic subtotal of pathogen counts.
Identify the internal manuscript discrepancy.
Explicit parser count
2,703
−19
Explicit site-specific positive or negative observations.
Map each discrepancy to source lines or categories.
Prepared observations
2,673
−30
Observations remaining after same-day reconciliation and sequence preparation.
Retain a deterministic removal or merge audit.
The analytical cohorts become progressively more specific
Observations by pathogen
Observations by anatomic site
CRE and stool observations dominate the dataset. This imbalance does not invalidate the pooled analyses, but it means that pathogen-site stratification, hierarchical pooling, and sparse-cell warnings are essential.
Pipeline completion and publication readiness remain separate
Audit source
Error
Warning
Information
Interpretation
Patient-file parser
0
0
5
No parser error is recorded, but five provenance notices remain.
Sequence preparation
8
0
14
Twenty-two preparation issues remain, including eight error-class entries.
Outcome parser
0
38
126
Endpoint transformations and provenance require review, although linkage is complete.
Practical implication: The models can run correctly while the data audit remains unfinished. Statistical execution, source reconciliation, and publication readiness must remain three separate statuses.
III. Four-State Operational MM: the primary interpretable model
The model directly encodes the surveillance rule
The scientific message is straightforward. Leaving P is difficult. One negative remains fragile because the next result is almost equally likely to progress or reset. Two negatives are much stronger evidence, but the third negative still performs a meaningful confirmation function.
Forward progression becomes more favorable as negative evidence accumulates
Progression, reset, and recurrence probabilities
Stage size and latent-colonization gradient
Transition
Count / denominator
Probability
Patient-clustered 95% interval
Interpretation
P → N1
299 / 1,782
16.78%
14.09%–19.82%
The first qualifying negative is the main bottleneck.
N1 → N2
153 / 275
55.64%
49.34%–61.31%
Slightly more than half progress to a second negative.
N1 → P
121 / 275
44.00%
38.44%–50.48%
One negative remains close to a positive reset.
N2 → C
106 / 145
73.10%
66.13%–80.84%
The third qualifying negative confirms C.
N2 → P
39 / 145
26.90%
19.16%–33.87%
Two negatives are strong evidence but remain incomplete.
C → P
29 / 69
42.03%
31.37%–52.48%
A later positive occurs after local clearance.
The operational stages also show a coherent biological-posterior gradient
Stage
Observations
Sequences
Mean pooled HMM P(colonized)
Positive-conditioned HMM P(colonized)
Interpretation
P
1,906
234
89.14%
92.53%
High posterior colonization probability.
N1
300
144
13.23%
25.50%
Substantial decline after the first negative.
N2
153
108
1.22%
2.96%
Posterior colonization becomes very low.
C
146
88
0.29%
0.94%
Lowest posterior colonization probability.
The directional coherence is informative, but it does not convert the biological HMM into a validated diagnostic test. Its emission probabilities remain model-based and its initial-state sensitivity remains substantial.
The uninterrupted clean-run probability and eventual clearance fraction answer different questions
Two distinct clearance estimands
IMMEDIATE CLEAN RUN
6.82%
Product of P → N1, N1 → N2, and N2 → C with no persistence, reset, or later attempt.
EVENTUALLY REACHED C
37.61%
Observed fraction of eligible sequences that reached C after persistence, resets, and repeated attempts.
The first number describes one uninterrupted path. The second describes eventual observed success across the entire sequence. The two values must never share the same endpoint label.
Recurrence depends strongly on the analytical unit and follow-up denominator
Recurrence estimates by denominator
Endpoint
Numerator
Denominator
Estimate
Meaning
Transition-level C → P
29
69 C-origin transitions
42.03%
Positive transition among observed transitions from C.
Followed cleared sequences
21
39 followed sequences
53.85%
At least one recurrence among sequences with follow-up.
All cleared sequences
21
88 cleared sequences
23.86%
Includes sequences without recurrence-detecting follow-up.
Followed cleared patients
17
27 followed patients
62.96%
At least one recurrent sequence among followed patients.
All cleared patients
17
43 cleared patients
39.53%
Includes patients without post-clearance surveillance.
Time to first recurrence is a separate endpoint. The median was 11 days, with an interquartile range of 7 to 31 days. A future recurrent-event model should explicitly incorporate time at risk and censoring.
IV. Four-State switching HMM: why two hidden regimes are preferred
The HMM does not replace P, N1, N2, and C
This model asks whether one transition matrix adequately describes all sequences. The answer appears to be no: the data are better described by two persistent transition patterns. However, these patterns remain statistical transition regimes and must not be relabeled as biological states or causal patient subtypes.
The two-regime model provides the best balance of fit, prediction, and parsimony
Model
Parameters
Converged
AICc
BIC
CV log loss
CV Brier
Minimum occupancy
Minimum separation
Four-State MM
6
Yes
2,254.700
2,288.967
0.506774
0.324643
100.00%
NE
HMM · 2 regimes
15
Yes
2,164.659
2,250.204
0.486243
0.312333
31.64%
34.13%
HMM · 3 regimes
26
Yes
2,173.920
2,321.938
0.483468
0.309986
27.59%
12.57%
HMM · 4 regimes
39
Yes
2,193.655
2,415.218
0.482243
0.308619
13.15%
10.74%
HMM · 5 regimes
54
No
2,224.257
2,530.284
0.483921
0.310013
5.15%
12.69%
Fit–prediction–complexity landscape
Transition fingerprints
The criterion winners are not identical
AICc WINNER
HMM · 2 regimes
2,164.659
BIC WINNER
HMM · 2 regimes
2,250.204
RAW CV LOG-LOSS WINNER
HMM · 4 regimes
0.482243
RAW CV BRIER WINNER
HMM · 4 regimes
0.308619
PRIMARY MODEL
Four-State MM
Direct protocol interpretation
RECOMMENDED HMM
HMM · 2 regimes
Best overall balance
The four-regime HMM lowers CV log loss by only 0.004 relative to the two-regime HMM. In exchange, it increases the parameter count from 15 to 39, worsens BIC by approximately 165 points, reduces minimum occupancy from 31.64% to 13.15%, and decreases minimum transition-profile separation from 34.13% to 10.74%.
Model-selection principle: A tiny improvement in one cross-validation split should not override complexity, convergence, occupancy, separation, entropy, and interpretability.
The two-regime transition profiles differ materially
Regime 1 · lower progression tendency
From / To
P
N1
N2
C
P
92.45%
7.55%
0%
0%
N1
71.20%
0.29%
28.51%
0%
N2
56.97%
0%
0.96%
42.07%
C
83.54%
0%
0%
16.46%
Regime 2 · higher progression tendency
From / To
P
N1
N2
C
P
45.66%
54.34%
0%
0%
N1
29.56%
0.67%
69.77%
0%
N2
20.49%
0%
0.21%
79.30%
C
38.55%
0%
0%
61.45%
Regime 1 is characterized by persistent positivity, frequent resets, and weak maintenance of C. Regime 2 is characterized by rapid progression through N1 and N2 and stronger maintenance of C. The difference is large enough to be scientifically interesting, but external validation is required before assigning biological labels.
The correct publication wording uses two complementary models
That one transition law explains all latent heterogeneity.
Complementary heterogeneity
Four-State HMM · 2 regimes
Two persistent transition profiles and improved held-out prediction.
That the regimes are biological states or treatment-response classes.
Raw predictive sensitivity
Four-State HMM · 4 regimes
The lowest single-split CV scores.
That it is the preferred overall model.
V. Other HMM families: related, but scientifically different
The application contains three distinct HMM questions
The biological HMM shows a coherent stage gradient but remains highly sensitive
Stage-posterior gradient across initial-state profiles
Initial-state profile BIC
Profile
BIC
P
N1
N2
C
Role
Positive-conditioned baseline
2,206.435
92.53%
25.50%
2.96%
0.94%
Design-aligned primary profile
Estimated initial state
2,187.042
86.54%
24.47%
3.38%
0.63%
Sensitivity profile
Stationary initial state
2,179.351
86.56%
24.67%
3.45%
0.63%
BIC-preferred sensitivity profile
Every profile produces a monotone P-to-C decline, but the alternative initial-state profiles have materially lower BIC than the design-aligned baseline. The model therefore supports a directional gradient while remaining too sensitive for strong biological claims.
The simple two-state HMM is better than its MM comparator but still inadequate
Criterion
Observed two-state MM
Two-state HMM
Relative result
Absolute adequacy result
AICc
≈ 2,578
≈ 2,508
HMM better by 70.642
Neither model adequate
BIC
≈ 2,596
≈ 2,537
HMM better by 58.932
CV log loss
0.498
0.485
HMM improves 2.53%
Predictive checks
2 of 4 flagged
4 of 4 flagged
HMM does not reproduce key features
A model may outperform another model and still be inadequate. Relative preference and absolute adequacy must remain separate conclusions.
VI. Outcome linkage and Death-inclusive modeling
Patient-level endpoint linkage is complete and methodologically structured
The continuous-time competing-risk model retains irregular observation time
A three-day interval and a fourteen-day interval do not contribute the same amount of time at risk. This is an important advantage over a purely discrete transition count. Exact event times remain interval-censored between cultures, so the homogeneous-rate approximation must remain visible.
Accumulated negative evidence raises the fitted probability of C before Death
Eventual absorption probabilities
Starting from P: fitted horizon probabilities
STARTING FROM P
42.07%
C before Death
95% interval 30.29%–52.89%
STARTING FROM N1
57.81%
C before Death
95% interval 45.77%–68.52%
STARTING FROM N2
77.73%
C before Death
95% interval 66.65%–85.57%
These are model-based first-event probabilities, not crude observed proportions and not validated release or treatment thresholds.
The 90-day result must display all three outcomes
10.59%
Confirmed C
23.41%
Death
66.00%
Still P, N1, or N2
Reporting only C or only Death would hide the fact that most episodes remain unresolved at 90 days when starting from P.
The current competing-risk model does not answer the discrete Five-State question
CURRENT REAL-DATA RESULT
Continuous-time competing risk
Exposure days are retained.
C and Death are competing first events.
Post-C recurrence is outside the first-event model.
Outputs include rates, absorption, and time horizons.
NEW RESULT STILL REQUIRED
Discrete Five-State MM
The next included transition is modeled directly.
Death is structurally absorbing.
C may remain C, recur to P, or proceed to Death.
The question is whether Death risk differs by source state.
Test whether added Death parameters justify complexity.
Not reported.
Bayes factor and posterior model probability
Quantify integrated structural evidence.
Not reported.
State-specific posterior Death intervals
Show uncertainty from P, N1, N2, and C.
Not reported.
Prior and linkage sensitivity
Test whether the verdict is robust.
Not reported.
VII. Terminal analyses: several target hypotheses are not supported
The prespecified negative-surge tests do not support the target hypothesis
Window and posterior contrasts
Observed weekly patient-weighted negativity
Analysis
Patients
Difference
95% interval
Probability or P value
Verdict
Final 4 versus previous 4 weeks
16 paired
−4.78 pp
−12.60 to +3.81
One-sided P = 0.8671
Increase not supported
Final 8 versus previous 8 weeks
10 paired
+3.85 pp
−11.98 to +20.94
One-sided P = 0.3355
Increase not supported
Death terminal versus earlier
28 paired
−8.53 pp
−14.08 to −2.69
Descriptive bootstrap interval
Lower near Death
Censoring terminal versus earlier
15 paired
−2.05 pp
−15.07 to +9.53
Descriptive bootstrap interval
Uncertain
The negative finding is scientifically valuable. It prevents an unsupported claim that negative cultures surge immediately before Death.
The exploratory change points are not supported
DEATH-ALIGNED
Boundary week 3
Earlier: 18.02%
Terminal: 13.28%
Difference: −4.74 pp
ΔBIC: −1.215
Bootstrap support: 23.60%
Not supported
CENSORING-ALIGNED
Boundary week 2
Earlier: 25.59%
Terminal: 36.19%
Difference: +10.59 pp
ΔBIC: −2.115
Bootstrap support: 37.80%
Not supported
The selected weeks may be retained as exploratory descriptive boundaries. They do not establish a biological switch or a terminal-illness mechanism.
The mixed model and GAMM prioritize different forms of evidence
Weekly adjusted predictions
Criterion
Mixed-effects logistic
GAMM
Winner
Fixed effects
14
24
Mixed is simpler
AICc
1,026.204
1,031.765
Mixed
BIC
1,099.624
1,153.620
Mixed
Patient-level CV log loss
0.645675
0.622606
GAMM
Shape flexibility
Linear endpoint trend
Nonlinear spline
GAMM
Current preference
More parsimonious
Preferred for prediction
GAMM under configured rule
The GAMM is preferred because it reduces patient-level CV log loss by 0.0231 while preserving a patient random intercept. The mixed model remains superior under AICc and BIC. This is a genuine trade-off rather than a contradiction.
The existing Bayesian terminal model quantifies uncertainty directly
Posterior contrasts and credible intervals
Weekly posterior predictions
Posterior contrast
Mean
95% credible interval
Posterior probability
Interpretation
Death terminal minus earlier
−2.24 pp
−8.19 to +3.58
P(increase) = 22.85%
No support for a death-terminal increase.
Censoring terminal minus earlier
+9.19 pp
+1.74 to +16.49
P(increase) = 99.33%
A censoring-aligned increase is highly probable under the model.
Death change minus censoring change
−11.44 pp
−20.99 to −1.76
P(> 0) = 1.10%
Death-aligned change is very unlikely to exceed censoring change.
This is a practical Bayesian advantage: uncertainty can be expressed as a direct posterior probability rather than only a binary significance decision. It also demonstrates that Bayesian analysis does not mechanically support the target hypothesis.
VIII. MLE and Bayesian estimation: different estimation methods, not competing model structures
MM versus HMM and MLE versus Bayesian are two separate axes
MLE provides the indispensable reference analysis
MLE contribution
Current use
Why it remains necessary
Direct transition estimates
Four-State MM probabilities
Transparent and directly auditable from transition counts.
Multi-start EM estimates
Hidden-regime HMM parameters
Provides a practical solution to latent-regime likelihood optimization.
Log likelihood
Model fit
Provides the basis for AICc and BIC.
AICc and BIC
Complexity-adjusted model comparison
Prevents unnecessary hidden-regime proliferation.
Grouped cross-validation
Held-out patient prediction
Provides an efficient predictive baseline.
Initialization
Future Bayesian fitting
A stable MLE solution can initialize posterior computation.
Bayesian estimation is realistic and useful only under specific conditions
①
Sparse-row stabilization
Posterior shrinkage can reduce unstable zero or one transition estimates.
②
Credible intervals
Each transition probability can be reported as a distribution rather than only a point estimate.
③
Hierarchical pooling
Sparse pathogen-site strata can borrow information without being treated as identical.
④
Posterior prediction
Predictions can integrate parameter uncertainty rather than relying on one fitted point.
These advantages are conceptually real, but usefulness must be demonstrated in the actual dataset. Bayesian estimation should not be retained merely because it produces credible intervals.
Bayesian predictive superiority requires a direct held-out test
Bayesian superiority should be claimed only when the estimate is positive, the patient-cluster interval excludes zero, calibration is not worse, and the conclusion remains stable under reasonable prior choices.
Structural evidence and predictive Bayesian value are separate conclusions
Bayesian prediction does not improve
Bayesian prediction improves
Five-State structure not supported
Retain the shared-Death structure and MLE baseline.
Retain the simpler structure but use Bayesian shrinkage for prediction.
Five-State structure supported
Use state-specific Death probabilities without claiming Bayesian predictive superiority.
Both state-specific Death structure and Bayesian prediction are supported.
IX. Publication-readiness and evidence-domain coverage
Analysis domain
Structure
Prediction
Uncertainty
Identifiability
Death handling
Current publication role
Four-State MM
Complete
Patient-level CV available
Patient-cluster bootstrap
Directly interpretable
Not primary
Primary model
Two-regime switching HMM
Complete
Log loss and Brier improved
Repeated-CV interval pending
Occupancy and separation acceptable
Not included
Complementary model
Biological HMM
Complete
Not primary purpose
Posterior summaries available
Highly initial-state sensitive
Not included
Exploratory
Simple two-state suite
Structurally inadequate
Predictive checks fail
Intervals available
Emission warning
Not included
Reference only
Competing-risk multi-state
Complete
Horizon probabilities
400 cluster bootstraps
Continuous-time approximation
C and Death first events
Supported with caution
Discrete Five-State MM
Architecture prepared
Planned
Posterior intervals planned
Count thresholds required
State-specific Death planned
Real-data result pending
Bayesian MM / HMM
Selected structure required first
Held-out comparison pending
Posterior framework possible
Prior sensitivity required
Depends on selected model
Utility untested
Claims that may and may not be made now
May be stated
The Four-State MM directly represents the three-negative operational process.
May be stated with caution
Two persistent transition regimes improve penalized fit and held-out prediction.
May be stated with caution
C-before-Death probability rises from P through N2 under the fitted generator.
Must not be stated
The four-regime HMM is the overall best model because one CV score is lowest.
Must not be stated
The hidden regimes are validated biological patient subtypes.
Must not be stated
A discrete Five-State MM or Bayesian prediction has already been proven superior.
X. What should be completed next?
The remaining work is concentrated and sequential
Priority table
Priority
Required task
Why it matters
Completion criterion
Critical
Resolve observation and sequence-preparation provenance
The source denominator remains under review.
Every difference is source-traceable and reproducible.
Critical
Run shared-Death versus Five-State comparison
The central fifth-state question remains unanswered.
Counts, fit, Bayes factor, posterior intervals, and sensitivity are populated.
Critical
Run MLE-versus-Bayesian predictive comparison
Bayesian usefulness must be demonstrated rather than assumed.
Held-out loss, calibration, cluster interval, and prior sensitivity are reported.
High
Repeated patient-level cross-validation
One fold allocation does not establish model-order stability.
The two-regime recommendation remains stable across assignments.
High
Paired patient-cluster bootstrap
Current predictive gains are point comparisons.
MM-versus-HMM log-loss and Brier intervals are available.
High
Calibration diagnostics
Average loss does not fully show probability reliability.
Reliability, calibration error, and sharpness are reviewed.
High
Canonical report integration
The new publication-evidence analyses are not yet in the current report.
Execution, persistence, visualizations, and copyable records are fully integrated.
XI. Final balanced verdict
Statement
Current verdict
Reason
The analytical pipeline is functioning.
Supported
All required analytical families completed and generated results.
The Four-State MM should remain the primary model.
Supported
It directly represents the protocol-defined P, N1, N2, and C process.
The two-regime switching HMM is the best-balanced complementary HMM.
Supported with caution
It balances AICc, BIC, prediction, occupancy, and separation.
The four-regime HMM is the overall best model.
Not supported
Its tiny raw CV gain does not justify its much greater complexity.
The biological HMM identifies validated biological states.
Not supported
The model is highly sensitive to its initial-state assumption.
The final pre-Death period shows a confirmed rise in negativity.
Not supported
The prespecified window contrasts and posterior results do not support the increase.
The competing-risk model is functioning.
Supported with caution
It provides coherent rates, absorption probabilities, and horizons under an approximation.
A discrete Five-State MM is superior.
Not yet tested in the canonical report
The required nested shared-Death comparison is absent.
Bayesian MM or HMM prediction is superior to MLE.
Not yet tested
No direct held-out predictive comparison has been reported.
Final conclusion: The project is functioning and the interpretation has become substantially more mature. The current publication strategy should not be a single-winner narrative. It should be a layered analytical portfolio: Four-State MM for primary interpretation, two-regime switching HMM for complementary transition heterogeneity and prediction, competing-risk analysis for confirmed clearance versus Death, and biological or terminal Bayesian models for carefully bounded exploratory questions. The remaining work is focused and concrete: close the data provenance, confirm HMM-order stability, run the discrete Five-State Death comparison, test whether Bayesian prediction truly improves upon MLE, and integrate those results into the canonical report.
현재 nGeneMDRO 분석은 제대로 작동하며, 전체 결과는 어떻게 해석해야 하는가?
종합 평가: 분석 pipeline은 작동하고 있으며, 모든 필수 분석 family가 완료되었고, 최신 보고서에는 이전보다 훨씬 타당한 model-selection 전략이 반영되어 있습니다. 현재 가장 강하게 지지되는 결론은 한 모델이 모든 다른 모델을 이겼다는 것이 아닙니다. 현재 근거는 layered model portfolio를 지지합니다. 즉, Four-State Operational MM은 직접 해석을 위한 주 모델, two hidden regimes를 가진 Four-State switching HMM은 가장 균형 잡힌 보완 HMM, continuous-time competing-risk model은 confirmed clearance와 Death의 경쟁 위험을 다루는 모델, biological HMM은 exploratory model입니다. Discrete Five-State Death 비교와 MLE-versus-Bayesian predictive comparison은 canonical report에 아직 없습니다. 따라서 전체 프로젝트는 잘 작동하고 있지만, 최종 publication-validation layer까지 완료된 상태는 아닙니다.
실행 상태
완료
Data pipeline: 3 / 3
Core analyses: 6 / 6
Terminal analyses: 11 / 11
Competing-risk analyses: 4 / 4
DATA AUDIT
검토 필요
2,772 → 2,722
2,722 → 2,703
2,703 → 2,673
Preparation error 8건
주 모델
Four-State MM
234개 sequence
91명 환자
2,505개 operational observation
직접 해석 가능
가장 균형 잡힌 HMM
Two hidden regimes
ΔAICc: +90.04
ΔBIC: +38.76
CV log-loss 개선: 4.05%
CV Brier 개선: 3.79%
DEATH ENDPOINT
완전 연결
98 / 98명
Death 44명
Independent censoring 37명
Chronology conflict 0명
COMPETING RISK
작동
199개 episode
77명 환자
C-first 54 · Death-first 83
Bootstrap 400 / 400
DISCRETE FIVE-STATE
결과 대기
Shared Death 대
State-specific Death
AICc · BIC · Bayes factor
Posterior Death interval
BAYESIAN MM / HMM
유용성 미검정
MLE 대 posterior prediction
Held-out log loss
Brier · calibration
Prior sensitivity
I. 직접적인 답변: 프로젝트는 제대로 작동하는가?
Analytical execution layer는 작동하고 있습니다
모든 필수 data-pipeline, core-analysis, terminal-analysis, competing-risk family가 완료되었습니다. Canonical report에는 29개의 analytical section이 포함되어 있고, required execution status는 Completed입니다. 이는 underlying analytical engine, model execution, result persistence, report assembly가 작동하고 있음을 강하게 보여줍니다.
Execution completeness
Completed
요청된 분석 family들이 실행을 마쳤음을 의미합니다.
Evidence quality
Mixed
Supported, exploratory, not supported 결과가 함께 존재합니다.
Report inclusion
29 sections
Documentation coverage를 의미하며 scientific validity를 자동으로 증명하지는 않습니다.
Prespecified 및 exploratory endpoint analysis가 실행되었습니다.
Target terminal hypothesis가 지지되었다는 뜻은 아닙니다.
Competing risk
4 / 4 completed
Death-inclusive exposure-time model이 작동합니다.
Discrete Five-State MM이 우수하다는 뜻은 아닙니다.
Optional diagnostics
0 / 1 completed
Optional diagnostic이 pending임을 명확히 보여줍니다.
Model-order stability가 완전히 확립되었다는 뜻은 아닙니다.
최신 model-selection 문구는 이전보다 더 과학적으로 타당합니다
이전 output은 two-regime HMM이 transition prediction에서 Four-State MM보다 지지된다는 점을 강조했습니다. 최신 output은 역할을 더 정확하게 분리합니다. Four-State MM은 primary protocol-aligned model로 유지하고, two-regime HMM은 transition prediction과 latent transition heterogeneity를 위한 supported complementary model로 표현합니다.
Decision component
이전 framing
현재 framing
개선된 이유
Primary model
HMM이 overall winner처럼 보일 수 있었습니다.
Four-State MM을 primary로 유지합니다.
Three-negative protocol의 직접 해석을 보존합니다.
HMM role
Four-State MM보다 지지됨.
Supported complementary model.
Prediction과 endpoint definition을 분리합니다.
Probability accuracy
CV log-loss improvement가 중심이었습니다.
CV log loss와 Brier improvement를 함께 표시합니다.
하나가 아니라 두 proper scoring rule을 사용합니다.
Universal winner
다소 모호할 수 있었습니다.
No universal single winner.
Interpretability와 prediction이 서로 다른 목적임을 반영합니다.
Mixed evidence는 실패가 아니라 적절한 결과입니다
Supported 또는 directly descriptive
Four-State transition, endpoint linkage, competing-risk cohort construction
Supported with caution
Two-regime switching HMM, GAMM, competing-risk probability
Final-four-week surge, final-eight-week elevation, terminal change point
Negative result와 uncertain result가 그대로 남아 있는 것이 오히려 report의 강점입니다. Publication-quality analytical system은 모든 결과를 positive conclusion으로 만들기보다 supported, exploratory, inconclusive, unsupported 상태를 분리해야 합니다.
II. Data foundation: linkage는 강하지만 provenance는 아직 미완료입니다
네 observation total은 서로 다른 stage입니다
Stage
Count
이전 stage와의 차이
의미
필요한 조치
Manuscript stated total
2,772
Reference
Manuscript에 기재된 전체 수입니다.
Source provenance로 유지합니다.
Manuscript pathogen subtotal
2,722
−50
Pathogen별 count의 arithmetic subtotal입니다.
Manuscript 내부 discrepancy를 확인합니다.
Explicit parser count
2,703
−19
Explicit site-specific positive 또는 negative observation입니다.
각 discrepancy를 source line 또는 category에 연결합니다.
Prepared observations
2,673
−30
Same-day reconciliation과 sequence preparation 이후 남은 observation입니다.
Deterministic removal 또는 merge audit를 유지합니다.
Analytical cohort는 단계적으로 더 구체화됩니다
Pathogen별 observation
Anatomic site별 observation
CRE와 stool observation이 dataset을 크게 차지합니다. Pooled analysis가 무효라는 뜻은 아니지만, pathogen-site stratification, hierarchical pooling, sparse-cell warning이 반드시 필요함을 의미합니다.
실질적 의미: Data audit가 미완료인 상태에서도 model은 정상적으로 실행될 수 있습니다. Statistical execution, source reconciliation, publication readiness는 별도 status로 유지해야 합니다.
III. Four-State Operational MM: 직접 해석을 위한 주 모델
Surveillance rule을 직접 encode합니다
Scientific message는 명확합니다. P를 벗어나는 과정이 어렵습니다. 음성 한 번은 다음 결과가 progression과 reset 사이에서 거의 비슷하게 나뉘기 때문에 여전히 불안정합니다. 음성 두 번은 훨씬 강한 근거이지만, 세 번째 음성은 여전히 의미 있는 confirmation 역할을 수행합니다.
Uninterrupted clean-run probability와 eventual clearance fraction은 다른 질문입니다
서로 다른 두 clearance estimand
IMMEDIATE CLEAN RUN
6.82%
Persistence, reset, later attempt가 없는 P → N1 → N2 → C product입니다.
EVENTUALLY REACHED C
37.61%
Persistence, reset, repeated attempt 이후 결국 C에 도달한 eligible sequence fraction입니다.
첫 수치는 uninterrupted path를 설명하고, 두 번째 수치는 전체 sequence에서의 eventual observed success를 설명합니다. 같은 endpoint label을 사용해서는 안 됩니다.
Recurrence는 analytical unit과 follow-up denominator에 따라 크게 달라집니다
Denominator별 recurrence estimate
Endpoint
Numerator
Denominator
Estimate
의미
Transition-level C → P
29
69 C-origin transitions
42.03%
C에서 관찰된 transition 중 positive transition입니다.
Followed cleared sequences
21
39 followed sequences
53.85%
Follow-up이 있는 sequence 중 recurrence 1회 이상입니다.
All cleared sequences
21
88 cleared sequences
23.86%
Recurrence detection follow-up이 없는 sequence도 포함됩니다.
Followed cleared patients
17
27 followed patients
62.96%
Followed patient 중 recurrent sequence 1개 이상입니다.
All cleared patients
17
43 cleared patients
39.53%
Post-clearance surveillance가 없는 patient도 포함됩니다.
Time to first recurrence는 별도 endpoint입니다. Median은 11일, interquartile range는 7–31일입니다. 향후 recurrent-event model은 time at risk와 censoring을 명시적으로 반영해야 합니다.
IV. Four-State switching HMM: 왜 two hidden regimes가 권장되는가?
HMM은 P, N1, N2, C를 대체하지 않습니다
이 model은 하나의 transition matrix가 모든 sequence를 충분히 설명하는지를 묻습니다. 현재 답은 그렇지 않은 것으로 보입니다. Data는 두 개의 persistent transition pattern으로 더 잘 설명됩니다. 그러나 이 pattern은 statistical transition regime이며 biological state 또는 causal patient subtype으로 이름 붙여서는 안 됩니다.
Two-regime model이 fit, prediction, parsimony의 가장 좋은 균형을 제공합니다
Model-selection 원칙: 한 번의 cross-validation split에서 나타난 아주 작은 개선이 complexity, convergence, occupancy, separation, entropy, interpretability를 모두 압도해서는 안 됩니다.
두 regime의 transition profile은 실질적으로 다릅니다
Regime 1 · 낮은 progression tendency
From / To
P
N1
N2
C
P
92.45%
7.55%
0%
0%
N1
71.20%
0.29%
28.51%
0%
N2
56.97%
0%
0.96%
42.07%
C
83.54%
0%
0%
16.46%
Regime 2 · 높은 progression tendency
From / To
P
N1
N2
C
P
45.66%
54.34%
0%
0%
N1
29.56%
0.67%
69.77%
0%
N2
20.49%
0%
0.21%
79.30%
C
38.55%
0%
0%
61.45%
Regime 1은 persistent positivity, frequent reset, weak C maintenance로 특징지어집니다. Regime 2는 N1과 N2를 빠르게 통과하고 C를 더 잘 유지합니다. 차이는 과학적으로 흥미롭지만 biological label을 붙이기 위해서는 external validation이 필요합니다.
모든 profile에서 P-to-C decline은 monotone합니다. 그러나 alternative initial-state profile의 BIC가 design-aligned baseline보다 훨씬 낮습니다. 따라서 directional gradient는 보고할 수 있지만 strong biological claim에는 적절하지 않습니다.
Simple two-state HMM은 MM보다 낫지만 여전히 inadequate합니다
Criterion
Observed two-state MM
Two-state HMM
Relative result
Absolute adequacy result
AICc
≈ 2,578
≈ 2,508
HMM better by 70.642
Neither model adequate
BIC
≈ 2,596
≈ 2,537
HMM better by 58.932
CV log loss
0.498
0.485
HMM improves 2.53%
Predictive checks
2 of 4 flagged
4 of 4 flagged
HMM이 key feature를 재현하지 못함
한 model이 다른 model보다 우수하더라도 두 model 모두 inadequate할 수 있습니다. Relative preference와 absolute adequacy는 별도 결론입니다.
3일 interval과 14일 interval이 같은 time at risk로 계산되지 않습니다. 이는 purely discrete transition count보다 중요한 장점입니다. Exact event time은 culture 사이에서 interval-censored되어 있으므로 homogeneous-rate approximation warning은 유지해야 합니다.
Negative finding도 scientific value가 있습니다. Death 직전에 negative culture가 급증한다는 unsupported claim이 manuscript에 들어가는 것을 방지합니다.
Exploratory change point는 지지되지 않았습니다
DEATH-ALIGNED
Boundary week 3
Earlier: 18.02%
Terminal: 13.28%
Difference: −4.74 pp
ΔBIC: −1.215
Bootstrap support: 23.60%
Not supported
CENSORING-ALIGNED
Boundary week 2
Earlier: 25.59%
Terminal: 36.19%
Difference: +10.59 pp
ΔBIC: −2.115
Bootstrap support: 37.80%
Not supported
선택된 week는 exploratory descriptive boundary로 유지할 수 있습니다. Biological switch 또는 terminal-illness mechanism을 증명하지는 않습니다.
Mixed model과 GAMM은 서로 다른 evidence를 우선합니다
Weekly adjusted prediction
Criterion
Mixed-effects logistic
GAMM
Winner
Fixed effects
14
24
Mixed가 더 단순함
AICc
1,026.204
1,031.765
Mixed
BIC
1,099.624
1,153.620
Mixed
Patient-level CV log loss
0.645675
0.622606
GAMM
Shape flexibility
Linear endpoint trend
Nonlinear spline
GAMM
Current preference
더 parsimonious함
Prediction에서 preferred
Configured rule에서 GAMM
GAMM은 patient random intercept를 유지하면서 patient-level CV log loss를 0.0231 낮추었기 때문에 preferred입니다. Mixed model은 AICc와 BIC에서 더 우수합니다. 이는 contradiction이 아니라 실제 trade-off입니다.
기존 Bayesian terminal model은 uncertainty를 직접 정량화합니다
Posterior contrast와 credible interval
Weekly posterior prediction
Posterior contrast
Mean
95% credible interval
Posterior probability
해석
Death terminal minus earlier
−2.24 pp
−8.19 to +3.58
P(increase) = 22.85%
Death-terminal increase를 지지하지 않습니다.
Censoring terminal minus earlier
+9.19 pp
+1.74 to +16.49
P(increase) = 99.33%
Model 안에서 censoring-aligned increase probability가 높습니다.
Death change minus censoring change
−11.44 pp
−20.99 to −1.76
P(> 0) = 1.10%
Death-aligned change가 censoring change보다 클 가능성이 매우 낮습니다.
이것이 Bayesian의 현실적인 장점입니다. Binary significance decision뿐 아니라 직접적인 posterior probability를 표현할 수 있습니다. 동시에 Bayesian analysis가 target hypothesis를 자동으로 지지하지 않는다는 점도 보여줍니다.
VIII. MLE와 Bayesian estimation: model structure가 아니라 estimation method의 차이
Approximation 아래에서 coherent rate, absorption probability, horizon을 제공합니다.
Discrete Five-State MM이 superior합니다.
Canonical report에서 아직 미검정
필요한 shared-Death nested comparison이 없습니다.
Bayesian MM 또는 HMM prediction이 MLE보다 superior합니다.
아직 미검정
Direct held-out predictive comparison이 보고되지 않았습니다.
최종 결론: 프로젝트는 작동하고 있으며 해석 구조도 상당히 성숙해졌습니다. 현재 publication strategy는 single-winner narrative가 아니라 layered analytical portfolio가 되어야 합니다. 즉, Four-State MM은 primary interpretation, two-regime switching HMM은 complementary transition heterogeneity와 prediction, competing-risk analysis는 confirmed clearance versus Death, biological 또는 terminal Bayesian model은 제한된 exploratory question에 사용해야 합니다. 남은 작업은 명확합니다. Data provenance를 마무리하고, HMM-order stability를 확인하며, discrete Five-State Death comparison을 실행하고, Bayesian prediction이 실제로 MLE보다 나은지를 검정한 뒤, 모든 결과를 canonical report에 통합해야 합니다.
Written on August 4, 2026
Interpreting the complete nGeneMDRO analysis: model hierarchy, evidence, and publication-ready conclusion (Written August 5, 2026)
The completed nGeneMDRO package now contains 36 canonical result sections, including the
Four-State operational analysis, biological and switching hidden Markov models, patient-specific surveillance
windows, terminal-outcome models, the P/N1/N2/C/Death competing-risk analysis, Publication Model Evidence,
and additional patient-cluster statistical diagnostics.
The overall evidence label is Mixed. This does not mean that the analysis failed. It means that
the different models answer different scientific questions and do not all support the same level of certainty.
A protocol model can be clearly supported as the primary explanatory framework while a more complicated
extension remains uncertain, exploratory, or useful only for prediction.
I. Executive conclusion
The central answer
The Four-State Operational MM should remain the primary model.
The Four-State switching HMM with two hidden transition regimes is the best-supported
secondary model for explaining additional transition heterogeneity and improving prediction.
The state-specific Five-State Death model is not established as superior, and Bayesian
posterior prediction is useful but not yet demonstrably better than plug-in MLE prediction.
There is therefore no defensible single-model-wins-everything conclusion. The appropriate conclusion is
hierarchical:
Primary explanatory model
Four-State Operational MM
Best alignment with the three-negative clearance protocol
Best-supported predictive extension
Four-State HMM with two hidden regimes
Better penalized fit and patient-separated prediction
Death-structure conclusion
Four-State shared Death versus Five-State Death
Mixed evidence; no stable replacement of the simpler structure
Outcome extension
P/N1/N2/C/Death competing-risk model
Useful for C-before-Death and horizon probabilities
Bayesian conclusion
Useful uncertainty and shrinkage layer
Predictive superiority remains inconclusive
Exploratory biological model
Clear-versus-Colonized biological HMM
Strong stage gradient but highly sensitive to initial-state assumptions
Which model is “best” depends on the question
Scientific question
Best current model
Current evidence
Recommended role
How does the three-negative rule operate?
Four-State Operational MM
Directly interpretable and statistically coherent
Primary analysis
Are there unobserved transition patterns beyond one common matrix?
Four-State HMM with two hidden regimes
Supported, with mild cross-validation disagreement
Secondary model
Should Death have different probabilities from P, N1, N2, and C?
No definitive winner
Mixed evidence
Sensitivity analysis
What is the probability of confirmed C before Death?
Continuous-time competing-risk multi-state model
Supported with caution
Outcome extension
Does Bayesian prediction outperform MLE?
No definitive winner
Inconclusive
Uncertainty and shrinkage adjunct
Does latent biological colonization probability decline across P, N1, N2, and C?
Positive-conditioned biological HMM
Strong gradient, exploratory model
Mechanistic supporting analysis
How does culture negativity change near Death or censoring?
GAMM for prediction; Bayesian model for probability statements
Supported with caution or exploratory
Terminal-outcome analysis
II. The model family must be separated before the results can be understood
One dataset is being examined through several different model lenses
Observed cultures
Positive / Negative
→
Operational history
P → N1 → N2 → C
→
Hidden transition regime
Lower- or higher-progression tendency
→
First-event outcome
Confirmed C or Death
These boxes do not represent one single model. They show a layered analytical architecture. The Four-State
MM reconstructs the observed protocol history. The switching HMM asks whether the same observed states behave
differently under latent transition regimes. The competing-risk model adds time at risk and competing
absorption into C or Death.
The most frequently confused models
Model
Observed state or outcome
Hidden component
Main question
Current role
Four-State Operational MM
P, N1, N2, C
None
What happens at the next included culture?
Primary
Biological two-state HMM
Positive or negative culture
Clear or Colonized
What latent biological state may have generated the culture?
Exploratory
Four-State switching HMM
P, N1, N2, C
One to five hidden transition regimes
Does the operational transition matrix change across latent regimes?
Secondary predictive model
Four-State plus shared Death
P, N1, N2, C, and terminal Death
None
Can one common Death probability be used across operational states?
Structural baseline
Five-State MM
P, N1, N2, C, Death
None
Should Death probability differ according to the current operational state?
Sensitivity analysis
Continuous-time competing-risk model
P, N1, N2 transient; C and Death absorbing
None
What is the time-dependent probability of C before Death?
Outcome extension
Bayesian hierarchical terminal model
Observed culture negativity near an endpoint
Patient random effects and posterior uncertainty
What is the posterior probability of a terminal change?
Exploratory inferential model
A “Four-State model with two hidden regimes” is not a six-state clinical model.
P, N1, N2, and C remain the four observed operational states. The two hidden regimes select between
different transition matrices. The computational joint state space is larger, but the clinical labels
remain unchanged.
The current Bayesian components are not yet a learned causal Bayesian network
Patient, pathogen, site, sampling→Current operational state St+Hidden regime Rt→Next state or Death
This is a useful probabilistic dependency map, but the completed report does not present a separately learned
causal Bayesian network. The implemented Bayesian components are transition-probability estimation,
posterior prediction, prior-sensitivity analysis, and a Bayesian hierarchical terminal model. They should not
be described as causal network discovery.
III. Data foundation and cohort integrity
The report is complete, but the source-data audit still requires attention
2,772
Manuscript stated total
→ −50 →
2,722
Pathogen subtotal
→ −19 →
2,703
Explicit parsed observations
→ −30 →
2,673
Prepared observations
Data level
Count
Meaning
Publication implication
Patients
98
Complete culture cohort
Stable patient denominator
Explicit observations
2,703
Parsed site-specific positive or negative rows
Must remain distinct from manuscript totals
Prepared sequences
321
Patient × pathogen × site chains
Primary sequence universe
Observed-positive Four-State sequences
234
Eligible for protocol-aligned reconstruction
Primary Four-State cohort
Hidden-regime comparison sequences
198
Met the minimum sequence-length requirement
Model-order cohort differs from the full Four-State cohort
Parser issues
5 information notices
No parser warning or error in the summary
Traceability still required
Sequence-preparation issues
22, including 8 errors
Deterministic reconciliation issues remain
Must be resolved or individually justified before final submission
The distinction between 2,772, 2,722, 2,703, and 2,673 is not a cosmetic reporting issue. Each number belongs
to a different provenance stage. A publication-quality manuscript should preserve all four counts and explain
every reduction.
Outcome linkage is strong, but the Death linkage used by the publication comparison is narrower
Outcome component
Count
Use
Culture patients linked to an endpoint
98 / 98
Complete endpoint provenance
Death endpoints
44
Death-aligned and competing-risk analyses
Independent censoring endpoints
37
Primary non-death comparator
Culture-defined endpoints
17
Excluded from the primary neutral-censoring comparator
Date conflicts
0
No chronology conflict in the linked cohort
Death patients linked for Publication Model Evidence
41 / 44
One Death transition per patient
Death patients with multiple eligible sequences
30
The sequence with the latest pre-death observation was selected
The one-sequence-per-Death rule avoids duplicating a patient-level Death endpoint, but it is also a modeling
choice. Sensitivity analyses using alternative linkage rules would strengthen the Death-state comparison.
IV. What the primary Four-State Operational MM shows
The first qualifying negative is the principal bottleneck
P Positive
16.78% →
N1 One negative
55.64% →
N2 Two negatives
73.10% →
C Confirmed clearance
Only 16.78% of next included cultures move from P to N1. Once a first qualifying negative has occurred,
progression becomes more likely. After N2, 73.10% confirm C, while 26.90% reset to P. This is the clearest
evidence that the third negative is not redundant.
Forward progression or maintenance versus persistence, reset, or recurrence at each operational stage.
The rare N1→N1 too-soon transition is omitted from the paired display.
The patient-cluster diagnostics refine the interpretation
Contrast
Difference
95% cluster-bootstrap interval
Holm-adjusted P
Interpretation
Remain P versus start N1
+66.44 pp in favor of remaining P
+60.78 to +71.65 pp
0.00600
Strong support for the P bottleneck
N1→N2 versus N1→P
+11.64 pp in favor of progression
+0.35 to +23.08 pp
0.09795
Directionally favorable but not familywise significant
N2→C versus N2→P
+46.21 pp in favor of C
+32.82 to +60.75 pp
0.00600
Strong support for the third-negative confirmation step
C→C versus C→P
+15.94 pp in favor of maintenance
−3.90 to +39.40 pp
0.14193
Maintenance and recurrence are not clearly separated
The overall monotone forward-progression pattern received 100% patient-cluster bootstrap support. However,
the N1 comparison did not survive Holm familywise adjustment. N1 should therefore remain interpreted as a
fragile intermediate evidence state rather than an established biological turning point.
Immediate clean progression, eventual clearance, and recurrence are different endpoints
Endpoint
Estimate
Correct meaning
Uninterrupted P→N1→N2→C product
6.82%
One clean run without persistence, reset, or a later attempt
Sequences eventually reaching C
88 / 234 = 37.61%
Clearance achieved at any later point
Transition-level C→P
29 / 69 = 42.03%
Positive next transition among observed transitions from C
Recurrence among followed cleared sequences
21 / 39 = 53.85%
Detected recurrence where post-clearance follow-up existed
Recurrence among all cleared sequences
21 / 88 = 23.86%
Includes sequences without a counted follow-up transition
Recurrence among followed cleared patients
17 / 27 = 62.96%
Patient-level detected recurrence with follow-up
Median observed time to first recurrence
11 days
Timing endpoint, not a recurrence probability
Forty-nine cleared sequences had no counted post-clearance transition. Recurrence estimates are therefore
highly dependent on surveillance continuation and must always retain their numerator, denominator, unit, and
follow-up requirement.
V. What the HMM results mean, and which HMM is preferred
The biological HMM has a strong stage gradient but weak model robustness
The positive-conditioned biological HMM estimates the probability of latent colonization as:
P 92.53% → N1 25.50% → N2 2.96% → C 0.94%.
Every adjacent decline remained supported after patient-cluster bootstrap and Holm adjustment.
Estimated latent-colonization probability across the operational stages. A logarithmic vertical scale is
used so that the N2 and C estimates remain visible.
The gradient is statistically coherent, but the model remains Exploratory because its
initial-state sensitivity is Highly Sensitive. The positive-conditioned profile had BIC
2206.435, whereas the stationary initial-state profile had BIC 2179.351, a difference of approximately
27.08 points. A strong posterior gradient does not erase uncertainty about the assumptions that produced it.
The biological HMM supports the directional interpretation that negative evidence corresponds to lower
estimated colonization probability. It does not yet provide a sufficiently robust biological state model
for primary publication claims or clinical release decisions.
The switching-HMM comparison supports two hidden transition regimes
Candidate
Parameters
Converged
AICc
BIC
Patient CV log loss
Minimum occupancy
Minimum separation
Four-State MM
6
Yes
2254.700
2288.967
0.506774
100.00%
NE
One hidden regime
6
Yes
2254.700
2288.967
0.506774
100.00%
NE
Two hidden regimes
15
Yes
2164.659
2250.204
0.486243
31.64%
34.13%
Three hidden regimes
26
Yes
2173.920
2321.938
0.483468
27.59%
12.57%
Four hidden regimes
39
Yes
2193.655
2415.218
0.482243
13.15%
10.74%
Five hidden regimes
54
No
2224.257
2530.284
0.483921
5.15%
12.69%
AICc and BIC penalties relative to the best model. Lower is better.
Patient-separated cross-validation log loss. Lower is better.
Why two regimes are preferred even though four regimes have the lowest CV loss
AICc and BIC both select two hidden regimes. The four-regime model has the lowest single-partition
cross-validation loss, but its advantage over the two-regime model is only 0.82% in the
diagnostic comparison. That small predictive difference is accompanied by:
A BIC penalty of approximately 165.01 points relative to the two-regime model.
Thirty-nine parameters instead of fifteen.
Minimum regime occupancy of 13.15% instead of 31.64%.
Minimum transition-profile separation of 10.74% instead of 34.13%.
Greater risk that a hidden regime represents unstable subdivision rather than reproducible structure.
The two-regime model is therefore the defensible balance of fit, prediction, parsimony, occupancy, and
interpretability. Repeated patient-level cross-validation is still required before the order can be regarded
as fully stable.
What the two hidden regimes appear to represent
Hidden regime
Occupancy
Progression score
Illustrative transition pattern
Permitted interpretation
Regime 1
68.36%
−0.116
P persistence 92.45%; N2→C 42.07%; C→P 83.54%
Lower clearance-progression tendency
Regime 2
31.64%
+0.514
P→N1 54.34%; N2→C 79.30%; C maintenance 61.45%
Higher clearance-progression tendency
These regimes may be called latent transition phenotypes or
lower- and higher-progression transition regimes. They should not be labeled as biological
subtypes, immune states, treatment-response classes, or patient phenotypes without external validation.
VI. Does adding Death make the Five-State MM better?
The structural comparison asks a narrower question than the competing-risk model
Four-State plus shared Death
P, N1, N2, and C retain their operational transitions. A single common Death probability is imposed
across all four source states.
P, N1, N2, C ── common qD ──→ Death
Five-State state-specific Death MM
Death is an absorbing fifth state, and P, N1, N2, and C may each have a different Death-transition
probability.
P→D, N1→D, N2→D, C→D estimated separately
This comparison does not ask whether Death is clinically important. It asks whether the limited Death
transitions justify three additional state-specific parameters.
The statistical criteria disagree
Criterion
Result
Direction
Interpretation
Nominal likelihood-ratio test
Statistic 11.239; df 3; P = 0.01050
Favors Five-State
The additional parameters improve in-sample likelihood
ΔAICc for Five-State
+5.192
Favors Five-State
Predictive-fit emphasis accepts the added complexity
ΔBIC for Five-State
−11.998
Favors shared-Death baseline
Stronger complexity penalty rejects the extra parameters
Exact log Bayes factor
−2.790
Favors shared-Death baseline
Integrated evidence is lower for Five-State
Bayes factor for Five-State
0.06139
Favors shared-Death baseline
Observed evidence is about 16 times more compatible with the baseline under the specified priors
Equal-prior posterior probability of Five-State
5.78%
Favors shared-Death baseline
Low posterior support at the primary prior scale
AICc and the nominal likelihood-ratio test reward the improved fit. BIC and the exact marginal-likelihood
calculation penalize the additional parameters more strongly. Because these criteria answer different
questions, the appropriate label is Mixed Evidence, not “Five-State wins.”
Prior sensitivity materially changes the Five-State probability
Equal-prior posterior probability of the Five-State structure under three prespecified prior scales.
Prior scale
P(Five-State)
Interpretation
0.5
28.23%
No material structural evidence
1.0
5.78%
Moderate evidence against Five-State
2.0
0.09%
Strong evidence against Five-State
A model conclusion that moves from 28.23% to 0.09% across reasonable prior scales is not robust enough to
support a definitive structural replacement.
The Death-transition counts explain much of the uncertainty
Transition
Observed count
Outgoing transitions
MLE
Posterior mean
95% posterior interval
P→Death
39
1,821
2.14%
2.17%
1.56% to 2.91%
N1→Death
1
276
0.36%
0.54%
0.04% to 1.70%
N2→Death
0
145
0.00%
0.34%
0.00% to 1.75%
C→Death
1
70
1.43%
2.11%
0.15% to 6.48%
Forty-one linked Death transitions are being distributed across four source states. Most occur from P,
while N1, N2, and C contain one, zero, and one event, respectively. The state-specific structure therefore
asks the data to estimate several sparse probabilities. This is exactly the setting in which AICc may favor
additional fit while BIC and Bayes factors favor restraint.
Practical conclusion for the Death-state question
The Five-State MM should be reported as a prespecified sensitivity model. It should not replace the
Four-State operational model, and the current evidence does not justify claiming that Death probability is
definitively state-specific.
The separate continuous-time competing-risk model remains valuable because it answers a different question:
the probability and timing of confirmed C versus Death while preserving exposure time in P, N1, and N2.
VII. What MLE and Bayesian analysis contributed
MLE provides the empirical transition fit
Maximum likelihood estimation uses the transition counts to identify the parameter values that make the
observed transitions most likely. It is transparent and efficient when transition counts are adequate.
Its main weakness in the current Five-State setting is that sparse cells can generate unstable estimates or
exact structural zeros, such as the observed N2→Death estimate of 0%.
Bayesian estimation adds shrinkage, intervals, and structural uncertainty
Bayesian estimation combines the observed transition counts with prespecified prior distributions. The
resulting posterior distribution provides:
Posterior means rather than only point estimates.
Credible intervals for sparse transition probabilities.
Posterior predictive probabilities for held-out transitions.
Marginal-likelihood evidence for comparing structural models.
Prior-sensitivity analysis showing whether conclusions depend heavily on prior scale.
The N2→Death cell illustrates the practical benefit. The MLE is exactly 0%, while the posterior mean is
0.34% with a 95% posterior interval from 0.00% to 1.75%. Bayesian estimation avoids treating the absence of an
observed event as proof that the event is impossible.
Bayesian prediction improved log loss slightly, but the result remains inconclusive
Patient-separated held-out multiclass log loss for plug-in MLE and Bayesian posterior prediction.
Lower is better.
Metric
Plug-in MLE
Bayesian
Improvement
95% patient-cluster interval
P(improvement)
Decision
Multiclass log loss
0.60805
0.58073
+0.02732
−0.00127 to +0.06357
95.25%
Inconclusive
Multiclass Brier score
0.34516
0.34513
+0.0000268
−0.0000685 to +0.0001184
71.55%
Inconclusive
The log-loss point estimate favors Bayesian prediction, but the patient-cluster interval still includes
zero improvement. The Brier scores are practically identical. Bayesian prediction may be modestly better,
but the current evidence does not establish superiority.
Bayesian analysis is already useful because it expresses uncertainty and stabilizes sparse probabilities.
Its value does not depend on proving that it always predicts better than MLE. Nevertheless, a claim of
predictive superiority is not supported by the current held-out evidence.
The Bayesian terminal model answers a different question
The Bayesian hierarchical terminal analysis is not the same as the Bayesian Five-State transition
comparison. It estimates terminal-minus-earlier changes in negative-culture probability while accounting for
repeated observations through a patient-level structure.
Posterior contrast
Mean
95% credible interval
Posterior probability of increase
Death terminal minus earlier
−2.24 pp
−8.19 to +3.58 pp
22.85%
Censoring terminal minus earlier
+9.19 pp
+1.74 to +16.49 pp
99.33%
Death change minus censoring change
−11.44 pp
−20.99 to −1.76 pp
1.10%
This result argues against a death-specific increase in culture negativity. The posterior distribution
instead places the death-aligned change below the censoring-aligned change.
VIII. Competing-risk and terminal-outcome interpretation
The competing-risk model adds clinically meaningful time-to-event context
The competing-risk cohort contains 199 patient-pathogen-site episodes from 77 patients:
54 confirmed-C-first episodes, 83 Death-first episodes, and 62 right-censored episodes. All 400 requested
patient-cluster bootstrap replicates were valid.
P⇄N1⇄N2→CorDeath
Eventual fitted probability of confirmed C before Death versus Death before confirmed C.
Accumulated negative evidence is associated with a progressively higher fitted probability of confirmed C
before Death: 42.07% from P, 57.81% from N1, and 77.73% from N2. This is one of the most clinically intuitive
findings, but it remains a model-based first-event probability rather than a validated release threshold.
Horizon probabilities show why eventual absorption is not the same as near-term outcome
Starting state
Horizon
Confirmed C
Death
Still P/N1/N2
P
30 days
1.32%
9.05%
89.63%
P
90 days
10.59%
23.41%
66.00%
P
365 days
35.67%
51.01%
13.32%
N1
90 days
34.13%
18.11%
47.76%
N2
90 days
65.25%
10.04%
24.71%
Starting from P, the eventual C-before-Death probability is 42.07%, but the 90-day probability of already
reaching confirmed C is only 10.59%. At that same horizon, 66.00% remain unresolved in P, N1, or N2. A report
that presents only eventual absorption would conceal the large near-term transient fraction.
The terminal analyses do not support a pre-Death increase in negative cultures
Bayesian posterior mean changes in patient-level negative-culture probability.
Analysis
Principal result
Interpretation
Final 4 versus previous 4 weeks
−4.78 pp; one-sided P = 0.8671
No supported final-four-week surge
Final 8 versus previous 8 weeks
+3.85 pp; one-sided P = 0.3355
Higher point estimate without confirmatory support
Death terminal versus earlier window
−8.53 pp; 95% bootstrap interval −14.08 to −2.69 pp
Lower negative fraction in the terminal window
Death change point
Week 3; ΔBIC −1.215; bootstrap support 23.60%
No supported abrupt boundary
Mixed-effects logistic trend
Death-versus-censoring trend ratio OR 0.634; 95% interval 0.434 to 0.927
Endpoint proximity behaves differently before Death
GAMM versus mixed logistic
CV log loss 0.623 versus 0.646
GAMM preferred for out-of-patient prediction
Bayesian terminal model
P(death-terminal increase) 22.85%
No posterior support for a death-terminal increase
The GAMM is preferred because it improves patient-level prediction, not because it proves a biological
terminal mechanism. Its BIC is worse than the simpler mixed model, and its spline coefficients are not
individually interpretable. The terminal findings remain observational.
IX. Integrated evidence map and publication hierarchy
Domain coverage map
The following map separates the domains in which each model performs well from domains that it was not
designed to answer.
Secondary evidence that one common transition matrix does not fully describe the operational process.
Main result 3
P/N1/N2/C/Death competing-risk model
Exposure-time transition rates, C-before-Death absorption, and horizon probabilities.
Sensitivity analysis
Shared-Death versus state-specific Five-State MM
Mixed evidence should be reported without a universal winner claim.
Methodological adjunct
MLE versus Bayesian posterior prediction
Bayesian shrinkage is useful, but predictive superiority remains inconclusive.
Exploratory supplement
Biological HMM and terminal models
Directional and predictive evidence with substantial assumption and identifiability limits.
A publication-ready integrated interpretation
The observed surveillance process is most transparently represented by the protocol-aligned Four-State
Operational MM. Transition progression becomes increasingly favorable after negative evidence accumulates,
with the principal bottleneck occurring at P→N1 and strong support for N2→C over reset. A Markov-switching
extension with two hidden transition regimes improves penalized fit and patient-separated prediction,
supporting latent transition heterogeneity while preserving P, N1, N2, and C as the observed states.
Evidence that Death requires state-specific discrete transition probabilities is mixed, and Bayesian
posterior prediction does not yet show conclusive held-out superiority over plug-in MLE. The continuous-time
competing-risk extension nevertheless provides informative model-based estimates of confirmed clearance
before Death and shows progressively higher C-before-Death probability from P through N2.
X. Claims that are supported and claims that should be avoided
Claims supported by the current results
The Four-State Operational MM is the primary protocol-aligned model.
P→N1 is the dominant operational bottleneck.
N2→C is substantially more likely than N2→P and remains supported after multiplicity adjustment.
The probability of forward progression increases as negative evidence accumulates.
A two-hidden-regime switching HMM is supported as a secondary transition model.
The two-regime HMM improves penalized fit and patient-separated prediction over the Four-State MM.
Confirmed-C-before-Death probability rises from P to N1 to N2 in the competing-risk model.
The final-four-week negative-culture surge hypothesis is not supported.
The final-eight-week increase has a positive point estimate but no confirmatory support.
Recurrence estimates vary materially according to the analytical unit and follow-up denominator.
Define a formal Bayesian network only after the nodes, temporal ordering, and causal versus predictive purpose are prespecified.
External validity
Perform temporal validation using a later surveillance period.
Perform external validation in another institution if possible.
Confirm whether the lower- and higher-progression regimes reproduce outside the original cohort.
Do not convert model probabilities into infection-control decisions until external calibration and decision analysis are complete.
XII. Final integrated answer
What should be regarded as the final model conclusion?
Rank
Model or analysis
Final interpretation
1
Four-State Operational MM
Primary model for explanation, protocol alignment, and the main manuscript narrative
2
Four-State HMM with two hidden regimes
Best-supported secondary model for latent transition heterogeneity and improved prediction
3
P/N1/N2/C/Death competing-risk model
Preferred outcome extension for time-aware C-before-Death inference
4
Five-State state-specific Death MM
Important sensitivity analysis, but mixed evidence prevents a superiority claim
5
Bayesian transition prediction
Useful for shrinkage and uncertainty; predictive advantage remains inconclusive
6
Biological HMM and terminal models
Exploratory supporting analyses rather than primary structural conclusions
Final answer: the current evidence does not support replacing the Four-State MM with one
universal alternative. The strongest publication structure is a layered model hierarchy:
Four-State MM as the primary protocol model, a two-hidden-regime Four-State HMM as the supported predictive
extension, and the competing-risk model as the Death-aware outcome extension. The Five-State Death structure
and Bayesian predictive advantage should be reported transparently as unresolved or sensitivity findings.
nGeneMDRO complete analysis 해석: 모델 위계, 근거 수준, 그리고 논문화 가능한 최종 결론
완성된 nGeneMDRO 결과 패키지는 현재 36개의 canonical result section을 포함합니다.
여기에는 4상태 운영적 분석, 생물학적 HMM과 숨은 전이 체제 HMM, 환자별 사망 전 감시 창 분석,
terminal-outcome model, P/N1/N2/C/Death 경쟁위험 분석, Publication Model Evidence,
그리고 환자 군집 기반 추가 통계진단이 모두 포함되어 있습니다.
전체 근거 수준은 Mixed입니다. 이는 분석이 실패했다는 뜻이 아닙니다.
서로 다른 모델이 서로 다른 연구 질문을 다루며, 각 질문에서 확보된 근거의 강도가 동일하지 않다는
의미입니다. 프로토콜을 설명하는 주 모델은 명확할 수 있지만, 더 복잡한 확장 모델은 예측용,
민감도 분석용 또는 탐색적 분석용으로 남을 수 있습니다.
I. 핵심 결론
가장 중요한 답
4상태 운영적 MM은 주 분석 모델로 유지되어야 합니다.2개의 숨은 전이 체제를 가진 4상태 switching HMM은 추가적인 전이 이질성을
설명하고 예측력을 개선하는 가장 근거가 강한 보조 모델입니다.
상태별 사망확률을 허용한 5상태 Death 모델이 더 우월하다는 결론은 확립되지 않았으며,
Bayesian posterior prediction은 유용하지만 plug-in MLE보다 확실히 우월하다고
입증되지는 않았습니다.
따라서 모든 질문에서 하나의 모델이 승리한다는 결론은 적절하지 않습니다. 현재 결과는 다음과 같은
위계적 결론을 지지합니다.
주 설명 모델
4상태 운영적 MM
3회 연속 음성이라는 프로토콜과 가장 직접적으로 일치
가장 근거가 강한 예측 확장
2개 숨은 체제의 4상태 HMM
복잡도 보정 적합도와 환자 분리 예측에서 개선
사망 구조 결론
공통 Death 확률 대 상태별 Five-State Death
근거가 혼재되어 단순 구조를 대체할 수 없음
결과 확장 모델
P/N1/N2/C/Death 경쟁위험 모델
C-before-Death 및 시점별 확률 산출에 유용
Bayesian 결론
불확실성과 수축을 제공하는 보조 계층
예측 우월성은 아직 불확실
탐색적 생물학 모델
Clear-versus-Colonized 생물학적 HMM
단계별 gradient는 강하지만 초기상태 가정에 매우 민감
“가장 좋은 모델”은 연구 질문에 따라 달라집니다
연구 질문
현재 가장 적절한 모델
현재 근거
권장 역할
3회 연속 음성 규칙이 실제로 어떻게 작동하는가?
4상태 운영적 MM
직접 해석 가능하며 통계적으로 일관됨
주 분석
하나의 공통 전이행렬로 설명되지 않는 숨은 전이 패턴이 있는가?
2개 숨은 체제의 4상태 HMM
경미한 CV 불일치를 동반한 지지
보조 모델
P, N1, N2, C마다 사망확률이 달라야 하는가?
확정적 승자 없음
Mixed Evidence
민감도 분석
사망 전에 확인된 C에 도달할 확률은 얼마인가?
연속시간 경쟁위험 다상태 모델
주의를 동반한 지지
결과 확장
Bayesian 예측이 MLE보다 우수한가?
확정적 승자 없음
Inconclusive
불확실성과 수축을 위한 보조 분석
P에서 C로 갈수록 잠재 집락화 확률이 감소하는가?
Positive-conditioned biological HMM
강한 gradient이나 모델 자체는 탐색적
기전적 보조 분석
사망 또는 censoring 근처에서 음성배양 비율이 어떻게 변하는가?
예측은 GAMM, 확률진술은 Bayesian model
주의를 동반한 지지 또는 탐색적
Terminal-outcome 분석
II. 결과를 이해하려면 먼저 모델들을 서로 분리해야 합니다
하나의 자료를 여러 모델 계층에서 분석하고 있습니다
관찰된 배양결과
Positive / Negative
→
운영적 이력
P → N1 → N2 → C
→
숨은 전이 체제
낮거나 높은 clearance 진행 경향
→
첫 사건 결과
확인된 C 또는 Death
이 도식의 상자들은 하나의 단일 모델을 뜻하지 않습니다. 서로 다른 분석 계층을 나타냅니다.
4상태 MM은 관찰된 프로토콜 이력을 재구성합니다. Switching HMM은 같은 P, N1, N2, C 상태가
숨은 전이 체제에 따라 다르게 움직이는지 평가합니다. 경쟁위험 모델은 P, N1, N2에 머문 시간과
C 또는 Death로의 흡수를 함께 다룹니다.
가장 자주 혼동되는 모델들의 차이
모델
관찰되는 상태 또는 결과
숨은 요소
주요 질문
현재 역할
4상태 운영적 MM
P, N1, N2, C
없음
다음 포함 배양에서 어떤 상태로 이동하는가?
주 모델
생물학적 2상태 HMM
Positive 또는 Negative culture
Clear 또는 Colonized
어떤 잠재 생물학 상태가 배양결과를 만들었는가?
탐색적
4상태 switching HMM
P, N1, N2, C
1–5개의 숨은 전이 체제
잠재 체제에 따라 운영적 전이행렬이 달라지는가?
보조 예측 모델
4상태 + 공통 Death
P, N1, N2, C 및 terminal Death
없음
모든 운영상태에 하나의 공통 사망확률을 적용할 수 있는가?
구조적 기준 모델
5상태 MM
P, N1, N2, C, Death
없음
현재 운영상태에 따라 사망확률이 달라야 하는가?
민감도 분석
연속시간 경쟁위험 모델
P, N1, N2는 transient; C와 Death는 absorbing
없음
시간에 따라 C가 Death보다 먼저 발생할 확률은 얼마인가?
결과 확장
Bayesian hierarchical terminal model
endpoint 근처의 관찰된 음성배양
환자 random effect 및 posterior uncertainty
terminal change의 posterior probability는 얼마인가?
탐색적 추론 모델
“2개의 숨은 체제를 가진 4상태 모델”은 6개의 임상상태를 가진 모델이 아닙니다.
P, N1, N2, C는 계속 4개의 관찰된 운영상태로 유지됩니다. 숨은 두 체제는 서로 다른 전이행렬을
선택하는 잠재 변수입니다. 계산상의 joint state space는 커지지만 임상상태의 명칭은 변하지 않습니다.
현재 Bayesian 구성은 아직 학습된 causal Bayesian network와는 다릅니다
환자·병원체·검체부위·검사빈도→현재 운영상태 St+숨은 체제 Rt→다음 상태 또는 Death
Prior + 관찰된 전이 → posterior transition probability → posterior predictive distribution
이 도식은 유용한 확률 의존성 지도이지만, 현재 complete report에는 별도로 학습된 인과적 Bayesian
network가 제시되어 있지 않습니다. 현재 구현된 Bayesian 요소는 전이확률의 사후추정, posterior
prediction, prior sensitivity, 그리고 Bayesian hierarchical terminal model입니다. 이를 causal
network discovery로 표현해서는 안 됩니다.
III. 자료 기반과 cohort의 정합성
결과 패키지는 완전하지만 원자료 audit은 아직 중요합니다
2,772
논문 기재 총계
→ −50 →
2,722
병원체별 소계
→ −19 →
2,703
명시적으로 파싱된 관찰
→ −30 →
2,673
준비된 관찰
자료 단계
수
의미
논문화 시 의미
환자
98
전체 배양 cohort
안정적인 환자 분모
명시적 관찰
2,703
검체부위별 positive 또는 negative row
논문 총계와 별도로 유지해야 함
준비된 sequence
321
환자 × 병원체 × 검체부위 chain
전체 sequence universe
Observed-positive 4상태 sequence
234
프로토콜 기반 재구성 대상
주 4상태 cohort
숨은 체제 비교 sequence
198
최소 sequence 길이 기준 충족
모델 차수 비교 cohort는 전체 cohort와 다름
Parser issue
정보성 notice 5개
요약상 parser warning 또는 error 없음
원문 추적성은 유지해야 함
Sequence-preparation issue
22개, 그중 error 8개
결정론적 reconciliation issue
최종 투고 전에 개별 검토 또는 정당화 필요
2,772, 2,722, 2,703, 2,673의 차이는 단순한 표기 문제가 아닙니다. 각 수치는 서로 다른 provenance
단계를 나타냅니다. 출판용 원고에서는 네 수치를 모두 남기고 각 감소의 근거를 설명해야 합니다.
전체 outcome linkage는 양호하지만 Publication Model Evidence의 Death linkage는 더 좁습니다
Outcome 구성
수
용도
Endpoint가 연결된 배양 환자
98 / 98
완전한 endpoint provenance
Death endpoint
44
사망 정렬 및 경쟁위험 분석
독립 censoring endpoint
37
주 비사망 비교군
배양으로 정의된 endpoint
17
중립 censoring 비교에서는 제외
날짜 충돌
0
연결 cohort에서 chronology conflict 없음
Publication Model Evidence에 연결된 Death 환자
41 / 44
환자당 하나의 Death transition
여러 eligible sequence를 가진 Death 환자
30
사망 전 가장 늦은 관찰이 있는 sequence 선택
환자당 하나의 sequence만 Death에 연결하는 규칙은 환자 수준 Death endpoint의 중복을 막지만,
동시에 하나의 모델링 선택입니다. 대안적 연결 규칙을 이용한 민감도 분석이 추가되면 사망상태 비교의
신뢰도가 높아질 수 있습니다.
IV. 주 분석인 4상태 운영적 MM이 보여주는 것
첫 번째 qualifying negative가 가장 큰 bottleneck입니다
P Positive
16.78% →
N1 음성 1회
55.64% →
N2 음성 2회
73.10% →
C 확인된 clearance
P에서 N1로 이동하는 다음 포함 배양은 16.78%에 불과합니다. 첫 qualifying negative가 발생한 뒤에는
진행 가능성이 높아집니다. N2 이후에는 73.10%가 C를 확인하고 26.90%가 P로 reset됩니다.
이는 세 번째 음성이 단순한 중복 검사가 아니라는 가장 분명한 근거입니다.
각 운영상태에서의 전진 또는 유지와, 지속·reset·재발의 비교입니다.
드문 N1→N1 too-soon transition은 이 paired display에서 제외했습니다.
환자 군집 통계진단은 해석을 더 엄격하게 만듭니다
대조
차이
95% cluster-bootstrap interval
Holm-adjusted P
해석
P 유지 대 N1 시작
P 유지가 +66.44 pp
+60.78 to +71.65 pp
0.00600
P bottleneck에 대한 강한 근거
N1→N2 대 N1→P
진행이 +11.64 pp
+0.35 to +23.08 pp
0.09795
방향은 유리하지만 familywise significance는 없음
N2→C 대 N2→P
C 진행이 +46.21 pp
+32.82 to +60.75 pp
0.00600
세 번째 음성 확인 단계에 대한 강한 근거
C→C 대 C→P
유지가 +15.94 pp
−3.90 to +39.40 pp
0.14193
유지와 재발이 명확히 분리되지 않음
전체적인 단조로운 forward-progression pattern은 100% patient-cluster bootstrap support를 보였습니다.
그러나 N1 대조는 Holm familywise adjustment를 통과하지 못했습니다. 따라서 N1은 생물학적 전환점이라기보다
여전히 불안정한 중간 evidence-history state로 해석하는 것이 적절합니다.
Clean progression, eventual clearance, recurrence는 서로 다른 endpoint입니다
Endpoint
추정치
올바른 의미
중단 없는 P→N1→N2→C product
6.82%
지속, reset, 재시도 없이 한 번에 진행한 경로
결국 C에 도달한 sequence
88 / 234 = 37.61%
후속 어느 시점이든 clearance 달성
Transition-level C→P
29 / 69 = 42.03%
C에서 관찰된 다음 전이 중 positive 전이
Follow-up이 있는 cleared sequence의 recurrence
21 / 39 = 53.85%
Post-clearance follow-up이 존재한 경우의 검출 재발
모든 cleared sequence의 recurrence
21 / 88 = 23.86%
추적 전이가 없는 sequence도 분모에 포함
Follow-up이 있는 cleared 환자의 recurrence
17 / 27 = 62.96%
환자 수준의 추적 관찰된 재발
첫 recurrence까지 관찰된 중앙값
11일
확률이 아닌 시간 endpoint
49개의 cleared sequence에는 post-clearance transition이 한 번도 집계되지 않았습니다.
따라서 recurrence 추정치는 감시가 얼마나 지속되었는지에 크게 의존하며, 항상 분자, 분모,
분석단위, follow-up 조건을 함께 표시해야 합니다.
V. HMM 결과의 의미와 어떤 HMM이 우선되는가
생물학적 HMM은 강한 stage gradient를 보이지만 모델 견고성은 부족합니다
Positive-conditioned biological HMM에서 잠재 집락화 확률은
P 92.53% → N1 25.50% → N2 2.96% → C 0.94%로 추정되었습니다.
인접한 모든 감소는 patient-cluster bootstrap과 Holm adjustment 이후에도 지지되었습니다.
Gradient 자체는 통계적으로 일관되지만, 모델은 Exploratory로 남습니다.
초기상태 민감도가 Highly Sensitive이기 때문입니다. Positive-conditioned profile의
BIC는 2206.435였지만 stationary initial-state profile의 BIC는 2179.351로 약 27.08 낮았습니다.
강한 posterior gradient가 그 gradient를 만든 가정의 불확실성을 제거하지는 못합니다.
생물학적 HMM은 음성 근거가 누적될수록 추정 집락화 확률이 감소한다는 방향성을 지지합니다.
그러나 현재 단계에서는 주 논문 결론이나 임상적 격리 해제 판단에 사용할 만큼 견고한 생물학적
상태 모델로 보기는 어렵습니다.
Switching-HMM 비교에서는 2개의 숨은 전이 체제가 지지됩니다
후보
Parameter
Converged
AICc
BIC
Patient CV log loss
최소 occupancy
최소 separation
4상태 MM
6
Yes
2254.700
2288.967
0.506774
100.00%
NE
숨은 체제 1개
6
Yes
2254.700
2288.967
0.506774
100.00%
NE
숨은 체제 2개
15
Yes
2164.659
2250.204
0.486243
31.64%
34.13%
숨은 체제 3개
26
Yes
2173.920
2321.938
0.483468
27.59%
12.57%
숨은 체제 4개
39
Yes
2193.655
2415.218
0.482243
13.15%
10.74%
숨은 체제 5개
54
No
2224.257
2530.284
0.483921
5.15%
12.69%
최적 모델 대비 AICc와 BIC penalty입니다. 낮을수록 좋습니다.
환자 분리 cross-validation log loss입니다. 낮을수록 좋습니다.
4개 체제의 CV가 조금 더 좋아도 2개 체제가 우선되는 이유
AICc와 BIC는 모두 2개 숨은 체제를 선택합니다. 4개 체제 모델은 한 번의 CV partition에서 가장 낮은
loss를 보였지만, 통계진단상 2개 체제와의 예측 차이는 0.82%에 불과합니다.
이 작은 예측 차이에는 다음 비용이 동반됩니다.
2개 체제 모델보다 약 165.01 높은 BIC penalty.
15개가 아닌 39개의 parameter.
31.64%가 아닌 13.15%의 최소 regime occupancy.
34.13%가 아닌 10.74%의 최소 transition-profile separation.
재현 가능한 구조가 아니라 불안정한 세분화를 포착했을 가능성.
따라서 2개 체제 모델이 적합도, 예측, 단순성, occupancy, 해석 가능성 사이의 가장 방어적인 균형입니다.
다만 모델 차수의 안정성을 확정하려면 반복 patient-level cross-validation이 필요합니다.
두 숨은 체제가 시사하는 전이 패턴
숨은 체제
Occupancy
Progression score
예시 전이 패턴
허용되는 해석
Regime 1
68.36%
−0.116
P 유지 92.45%; N2→C 42.07%; C→P 83.54%
낮은 clearance 진행 경향
Regime 2
31.64%
+0.514
P→N1 54.34%; N2→C 79.30%; C 유지 61.45%
높은 clearance 진행 경향
이 체제들은 latent transition phenotype 또는
낮은 진행 체제와 높은 진행 체제라고 표현할 수 있습니다.
외부 검증 없이 생물학적 subtype, 면역상태, 치료반응군, 환자 phenotype으로 명명해서는 안 됩니다.
VI. Death를 추가한 5상태 MM이 더 좋은가
이 구조 비교는 경쟁위험 모델보다 더 좁은 질문을 다룹니다
4상태 + 공통 Death
P, N1, N2, C의 운영적 전이는 유지됩니다. 네 출발상태 모두에 하나의 공통 Death 확률을 적용합니다.
P, N1, N2, C ── 공통 qD ──→ Death
상태별 Death를 가진 5상태 MM
Death를 흡수 5번째 상태로 두고 P, N1, N2, C 각각에서 서로 다른 Death transition probability를
허용합니다.
P→D, N1→D, N2→D, C→D를 별도 추정
이 비교는 Death가 임상적으로 중요한지를 묻는 것이 아닙니다. 제한된 Death transition으로
세 개의 추가 상태별 parameter를 정당화할 수 있는지를 묻습니다.
통계 기준들이 서로 다른 결론을 냅니다
기준
결과
방향
해석
Nominal likelihood-ratio test
Statistic 11.239; df 3; P = 0.01050
Five-State 지지
추가 parameter가 in-sample likelihood를 개선
Five-State ΔAICc
+5.192
Five-State 지지
예측 적합도 관점에서 복잡도 증가를 수용
Five-State ΔBIC
−11.998
공통 Death 기준 모델 지지
강한 복잡도 penalty가 추가 parameter를 거부
Exact log Bayes factor
−2.790
공통 Death 기준 모델 지지
Five-State의 integrated evidence가 더 낮음
Five-State Bayes factor
0.06139
공통 Death 기준 모델 지지
지정된 prior 아래에서 기준 모델 쪽 근거가 약 16배 큼
Equal-prior P(Five-State)
5.78%
공통 Death 기준 모델 지지
Primary prior scale에서 낮은 posterior support
AICc와 nominal likelihood-ratio test는 향상된 적합도를 보상합니다. BIC와 exact marginal likelihood는
추가 parameter를 더 강하게 벌점화합니다. 이 기준들은 서로 다른 질문에 답하므로 적절한 결론은
Mixed Evidence이며 “Five-State가 승리했다”가 아닙니다.
Prior sensitivity에 따라 Five-State 확률이 크게 변합니다
세 가지 prespecified prior scale에서의 Five-State 구조 posterior probability입니다.
Prior scale
P(Five-State)
해석
0.5
28.23%
실질적인 구조 근거 없음
1.0
5.78%
Five-State에 반대하는 중등도 근거
2.0
0.09%
Five-State에 반대하는 강한 근거
합리적인 prior scale 범위에서 모델 확률이 28.23%에서 0.09%까지 변한다면 구조적 대체를 확정할 만큼
견고한 결론으로 보기 어렵습니다.
Death-transition 수가 적다는 점이 불확실성을 설명합니다
Transition
관찰 수
출발상태 전이 수
MLE
Posterior mean
95% posterior interval
P→Death
39
1,821
2.14%
2.17%
1.56% to 2.91%
N1→Death
1
276
0.36%
0.54%
0.04% to 1.70%
N2→Death
0
145
0.00%
0.34%
0.00% to 1.75%
C→Death
1
70
1.43%
2.11%
0.15% to 6.48%
연결된 41개의 Death transition을 네 출발상태에 나누어 추정하고 있습니다. 대부분은 P에서 발생하고,
N1, N2, C에는 각각 1건, 0건, 1건만 있습니다. 상태별 사망구조는 여러 개의 sparse probability를
추정해야 합니다. 이런 상황에서는 AICc가 추가 적합도를 선호하면서 BIC와 Bayes factor가 단순성을
선호하는 결과가 자연스럽게 발생할 수 있습니다.
Death-state 질문에 대한 실무적 결론
5상태 MM은 prespecified sensitivity model로 보고하는 것이 적절합니다. 4상태 운영적 모델을
대체해서는 안 되며, 현재 자료만으로 사망확률이 운영상태별로 확실히 다르다고 주장할 수 없습니다.
별도의 연속시간 경쟁위험 모델은 여전히 유용합니다. 그 모델은 P, N1, N2의 exposure time을 보존하면서
확인된 C와 Death 중 어떤 사건이 먼저 발생하는지와 그 시점을 다루기 때문입니다.
VII. MLE와 Bayesian 분석이 각각 기여한 것
MLE는 관찰자료에 가장 잘 맞는 전이확률을 제공합니다
Maximum likelihood estimation은 관찰된 전이를 가장 가능하게 만드는 parameter를 찾습니다.
전이 수가 충분할 때 투명하고 효율적입니다. 그러나 현재 5상태 구조처럼 sparse cell이 존재하면
불안정한 추정치나 정확한 0이 발생할 수 있습니다. N2→Death의 관찰 MLE 0%가 대표적입니다.
Bayesian 추정은 shrinkage, interval, 구조적 불확실성을 추가합니다
Bayesian 추정은 관찰된 전이 수와 prespecified prior를 결합합니다. 그 결과 다음 정보를 제공합니다.
점추정치만이 아니라 posterior mean.
Sparse transition probability에 대한 credible interval.
Held-out transition에 대한 posterior predictive probability.
구조 모델 비교를 위한 marginal-likelihood evidence.
결론이 prior scale에 얼마나 의존하는지를 보여주는 sensitivity analysis.
N2→Death cell은 실질적인 장점을 잘 보여줍니다. MLE는 정확히 0%지만 posterior mean은 0.34%이며
95% posterior interval은 0.00%–1.75%입니다. 관찰사건이 없다는 이유만으로 사건이 불가능하다고
단정하는 것을 피할 수 있습니다.
Log-loss point estimate는 Bayesian을 선호하지만 patient-cluster interval이 여전히 0 improvement를
포함합니다. Brier score는 사실상 동일합니다. Bayesian prediction이 조금 더 좋을 가능성은 있으나,
현재 근거로 우월성을 확정할 수는 없습니다.
Bayesian 분석은 sparse probability를 안정화하고 불확실성을 정량화한다는 점에서 이미 유용합니다.
반드시 MLE보다 항상 예측을 잘해야만 가치가 있는 것은 아닙니다. 다만 현재 held-out 결과로 Bayesian
예측 우월성을 주장해서는 안 됩니다.
Bayesian terminal model은 별도의 질문에 답합니다
Bayesian hierarchical terminal analysis는 Bayesian Five-State transition 비교와 다른 분석입니다.
환자 내 반복관찰을 고려하면서 terminal period와 earlier period의 음성배양 확률 차이를 추정합니다.
Posterior contrast
Mean
95% credible interval
증가 posterior probability
Death terminal minus earlier
−2.24 pp
−8.19 to +3.58 pp
22.85%
Censoring terminal minus earlier
+9.19 pp
+1.74 to +16.49 pp
99.33%
Death change minus censoring change
−11.44 pp
−20.99 to −1.76 pp
1.10%
이 결과는 사망에 특이적인 음성배양 증가를 지지하지 않습니다. 오히려 posterior distribution은
death-aligned change가 censoring-aligned change보다 낮은 쪽에 위치합니다.
VIII. 경쟁위험 및 terminal-outcome 결과의 해석
경쟁위험 모델은 임상적으로 이해하기 쉬운 time-to-event 정보를 추가합니다
경쟁위험 cohort에는 77명의 환자로부터 나온 199개의 patient-pathogen-site episode가 포함되었습니다.
확인된 C가 먼저인 episode 54개, Death가 먼저인 episode 83개, right-censored episode 62개였으며,
요청한 400회의 patient-cluster bootstrap이 모두 유효했습니다.
P⇄N1⇄N2→C또는Death
확인된 C가 Death보다 먼저 발생할 최종 확률과 Death가 먼저 발생할 확률입니다.
음성 근거가 누적될수록 확인된 C가 Death보다 먼저 발생할 fitted probability가 높아집니다.
P에서는 42.07%, N1에서는 57.81%, N2에서는 77.73%입니다. 임상적으로 매우 직관적인 결과이지만,
검증된 격리 해제 기준이 아니라 model-based first-event probability입니다.
Horizon probability는 eventual absorption과 단기 결과가 다름을 보여줍니다
시작상태
Horizon
확인된 C
Death
여전히 P/N1/N2
P
30일
1.32%
9.05%
89.63%
P
90일
10.59%
23.41%
66.00%
P
365일
35.67%
51.01%
13.32%
N1
90일
34.13%
18.11%
47.76%
N2
90일
65.25%
10.04%
24.71%
P에서 시작한 eventual C-before-Death probability는 42.07%지만, 90일 이내에 이미 확인된 C에 도달할
확률은 10.59%입니다. 같은 시점에 66.00%는 P, N1, N2 중 하나에 아직 남아 있습니다.
Eventual absorption만 제시하면 큰 단기 미해결 transient fraction이 가려질 수 있습니다.
Terminal 분석은 사망 전 음성배양 증가를 지지하지 않습니다
환자 수준 음성배양 확률 변화의 Bayesian posterior mean입니다.
분석
주 결과
해석
마지막 4주 대 이전 4주
−4.78 pp; one-sided P = 0.8671
마지막 4주의 음성 surge는 지지되지 않음
마지막 8주 대 이전 8주
+3.85 pp; one-sided P = 0.3355
Point estimate는 높지만 confirmatory support 없음
Death terminal 대 earlier window
−8.53 pp; 95% bootstrap interval −14.08 to −2.69 pp
Terminal window에서 음성 비율이 더 낮음
Death change point
3주; ΔBIC −1.215; bootstrap support 23.60%
뚜렷한 급격한 경계는 지지되지 않음
Mixed-effects logistic trend
Death-versus-censoring trend ratio OR 0.634; 95% interval 0.434 to 0.927
Endpoint proximity pattern이 Death와 censoring에서 다름
GAMM 대 mixed logistic
CV log loss 0.623 대 0.646
Out-of-patient prediction에서는 GAMM 우선
Bayesian terminal model
P(death-terminal increase) 22.85%
사망 전 terminal increase에 대한 posterior support 없음
GAMM이 우선된 이유는 patient-level prediction이 개선되었기 때문이지 생물학적 terminal mechanism을
입증했기 때문이 아닙니다. GAMM의 BIC는 단순한 mixed model보다 더 나쁘며 spline coefficient는
개별적으로 해석할 수 없습니다. Terminal 결과는 관찰연구적 결과로 남습니다.
IX. 통합 근거 지도와 논문 내 모델 위계
Domain coverage map
다음 표는 각 모델이 잘 다루는 영역과 애초에 답하도록 설계되지 않은 영역을 구분합니다.
모델
프로토콜 해석성
복잡도 보정 적합도
환자 수준 예측
불확실성 정량화
Death 모델링
권장 지위
4상태 운영적 MM
강함
기준
기준
Patient-cluster bootstrap
주 목적 아님
주 모델
2개 체제의 4상태 HMM
중간
최저 AICc와 BIC
개선
Occupancy, entropy, separation
포함하지 않음
가장 좋은 보조 확장
상태별 Death 5상태 MM
중간
AICc 지지, BIC 반대
확정적이지 않음
Posterior interval과 Bayes factor
직접적
민감도 모델
Bayesian Five-State prediction
중간
같은 구조
소폭 개선, 불확실
강함
직접적
보조 분석
경쟁위험 다상태 모델
결과 해석이 강함
다른 likelihood target
Model-based horizon
Patient-cluster interval
직접적이며 time-aware
결과 확장
Biological Clear/Colonized HMM
간접적
상대 적합도는 유리
절대 adequacy 불확실
Posterior probability
주 목적 아님
탐색적
Terminal GAMM / Bayesian model
Clearance rule model 아님
기준이 혼재
CV에서는 GAMM 우선
Posterior probability 제공
Endpoint-relative 분석
탐색적 outcome analysis
권장 논문 구성
주 결과 1
4상태 운영적 MM
프로토콜 재구성, bottleneck, 전진, reset, 확인된 C, 분모별 recurrence.
관찰된 감시과정은 프로토콜과 직접 일치하는 4상태 운영적 MM으로 가장 투명하게 표현됩니다.
음성 근거가 누적될수록 전진 가능성이 증가하며, 가장 큰 bottleneck은 P→N1이고 N2→C는 reset보다
강하게 지지됩니다. 두 개의 숨은 전이 체제를 가진 Markov-switching 확장은 P, N1, N2, C를 관찰상태로
유지하면서 복잡도 보정 적합도와 환자 분리 예측을 개선하여 잠재적인 전이 이질성을 지지합니다.
반면 Death가 운영상태별로 서로 다른 discrete transition probability를 가져야 한다는 근거는 혼재되어
있으며, Bayesian posterior prediction이 plug-in MLE보다 held-out prediction에서 확실히 우월하다는
근거도 충분하지 않습니다. 다만 연속시간 경쟁위험 확장은 Death 이전에 확인된 C에 도달할 확률을
시간과 함께 제시하며, P에서 N2로 갈수록 C-before-Death probability가 높아지는 패턴을 보여줍니다.
X. 현재 근거로 가능한 주장과 피해야 할 주장
현재 결과가 지지하는 주장
4상태 운영적 MM은 프로토콜에 부합하는 주 모델입니다.
P→N1은 가장 큰 운영적 bottleneck입니다.
N2→C는 N2→P보다 현저히 높으며 multiplicity adjustment 이후에도 지지됩니다.
음성 근거가 누적될수록 forward progression probability가 증가합니다.
2개 숨은 체제 switching HMM은 보조 전이 모델로 지지됩니다.
2개 체제 HMM은 4상태 MM보다 penalized fit과 patient-separated prediction을 개선합니다.
경쟁위험 모델에서 C-before-Death probability는 P에서 N1, N2로 갈수록 증가합니다.
마지막 4주의 음성배양 surge 가설은 지지되지 않습니다.
마지막 8주는 높은 point estimate가 있으나 confirmatory support는 없습니다.
Recurrence 추정치는 분석단위와 follow-up 분모에 따라 크게 달라집니다.
Bayesian 추정은 sparse transition을 안정화하고 유용한 uncertainty interval을 제공합니다.
현재 결과가 지지하지 않는 주장
5상태 MM이 공통 Death 기준 모델보다 확실히 우월합니다.
Bayesian posterior prediction이 MLE보다 확실히 우월합니다.
두 숨은 체제가 검증된 생물학적 subtype입니다.
생물학적 HMM이 실제 Clear와 Colonized 상태를 견고하게 규명했습니다.
Terminal illness가 관찰된 배양음성 패턴을 유발했습니다.
확인된 local C가 검증된 치료, 격리 해제 또는 감염관리 기준입니다.
HMM emission sensitivity와 specificity가 외부 검증된 검사 성능과 같습니다.
C→P가 환자 수준 recurrence incidence와 같습니다.
검체 미채취 또는 채취 불가능을 negative culture로 간주할 수 있습니다.
XI. 최종 논문화 전에 우선해야 할 작업
자료와 provenance
논문 총계 내부의 50건 arithmetic gap을 해결하거나 명시적으로 설명해야 합니다.
19건의 manuscript-to-parser 차이를 source row 또는 category까지 추적해야 합니다.
30건의 parser-to-preparation 감소를 same-day duplicate 또는 conflict rule과 연결해야 합니다.
8개의 sequence-preparation error를 개별 검토해야 합니다.
전체 환자 98명, primary endpoint 환자 81명, competing-risk 환자 77명의 차이를 유지해야 합니다.
숨은 체제의 안정성
여러 fold assignment와 random seed로 patient-level cross-validation을 반복해야 합니다.
한 번의 fold 결과가 아니라 선택된 hidden-regime count의 분포를 보고해야 합니다.
환자당 하나의 sequence를 선택하는 다른 Death linkage rule로 분석을 반복해야 합니다.
최대 372일의 Death-linkage gap이 결과에 미치는 영향을 평가해야 합니다.
연령, 병원체, 검체부위, 검사빈도, 임상중증도를 포함한 adjusted state-specific Death model을 고려해야 합니다.
상태별 Death count가 sparse하면 hierarchical partial pooling이 필요합니다.
현재 homogeneous continuous-time approximation과 interval-censored 또는 semi-Markov formulation을 비교해야 합니다.
Bayesian 분석을 현실적으로 더 결정적으로 만드는 방법
독립적인 sparse transition row 대신 fully hierarchical Bayesian transition model을 적용해야 합니다.
의미 있는 이질성은 보존하면서 병원체, 검체부위, 운영상태 사이에 partial pooling을 적용해야 합니다.
Patient random-effect variance를 하나의 추정값에 고정하지 말고 그 불확실성도 전파해야 합니다.
여러 fold partition에서 patient-separated posterior predictive validation을 반복해야 합니다.
Calibration, log loss, Brier score, 임상적으로 의미 있는 decision consequence를 함께 보고해야 합니다.
정식 Bayesian network는 node, 시간순서, causal 또는 predictive 목적을 먼저 prespecify한 뒤 구축해야 합니다.
외부 타당도
더 이후 시기의 surveillance data를 이용한 temporal validation이 필요합니다.
가능하다면 다른 기관의 자료를 이용한 external validation이 필요합니다.
낮은 진행 체제와 높은 진행 체제가 원래 cohort 밖에서도 재현되는지 확인해야 합니다.
External calibration과 decision analysis가 완료되기 전에는 모델 확률을 감염관리 결정기준으로 전환해서는 안 됩니다.
XII. 최종 통합 답
최종 모델 결론을 어떻게 정리해야 하는가
순위
모델 또는 분석
최종 해석
1
4상태 운영적 MM
설명, 프로토콜 정합성, 주 논문 narrative를 위한 primary model
2
2개 숨은 체제의 4상태 HMM
잠재 전이 이질성과 개선된 예측을 위한 가장 근거가 강한 secondary model
3
P/N1/N2/C/Death 경쟁위험 모델
시간을 고려한 C-before-Death 추론을 위한 outcome extension
4
상태별 Death 5상태 MM
중요한 민감도 분석이지만 Mixed Evidence로 인해 우월성 주장 불가
5
Bayesian transition prediction
Shrinkage와 uncertainty에는 유용하지만 예측 장점은 불확실
6
Biological HMM 및 terminal model
주 구조 결론이 아니라 탐색적 supporting analysis
최종 답: 현재 근거는 4상태 MM을 하나의 보편적 대안으로 대체하는 결론을 지지하지
않습니다. 가장 강한 논문 구조는 layered model hierarchy입니다.
4상태 MM을 주 프로토콜 모델로 두고, 2개 숨은 체제의 4상태 HMM을 지지되는 예측 확장으로,
경쟁위험 모델을 Death-aware outcome extension으로 배치하는 구성이 적절합니다.
Five-State Death 구조와 Bayesian predictive advantage는 해결되지 않은 민감도 결과로 투명하게
보고하는 것이 가장 방어적입니다.
Written on August 5, 2026
Interpreting the complete nGeneMDRO analysis: a visual evidence atlas for MM, HMM, Death, MLE, and Bayesian models (Written August 5, 2026)
Canonical report
36 sections
Complete result package
Primary model
Four-State MM
Protocol explanation
Best secondary model
Two-regime HMM
Transition heterogeneity
Death structure
Mixed evidence
Five-State not established
Bayesian prediction
Inconclusive
Useful uncertainty layer
Overall evidence
Mixed
Different questions, different answers
The central finding is not that one model wins every comparison.
The defensible conclusion is a layered model hierarchy: the Four-State Operational MM for protocol explanation,
the two-hidden-regime Four-State HMM for additional predictive structure, and the continuous-time competing-risk
model for Death-aware outcome probabilities.
Green · supported or primaryBlue · descriptive or informativeAmber · caution or mixedRed · not supported or unstableGray · exploratory or not designed for that domain
I. Executive conclusion
The model hierarchy at one glance
Observed protocol history
Four-State MM
P → N1 → N2 → C
Positive resets
Post-C recurrence
Primary explanatory model
The primary model is extended, not replaced
Two-regime Four-State HMM
Adds latent transition heterogeneity while preserving P, N1, N2, and C.
Competing-risk multi-state model
Adds exposure time and competing absorption into C or Death.
Five-State Death sensitivity
Tests whether Death probability must differ by operational state.
Bayesian evidence layer
Adds shrinkage, posterior intervals, prediction, and prior sensitivity.
Exploratory mechanisms
Biological HMM and terminal models
Clear versus Colonized
Death-aligned patterns
GAMM and Bayesian trends
Final hierarchy: Four-State MM first, two-regime HMM second, competing-risk model as the
Death-aware extension, Five-State Death as a sensitivity analysis, and Bayesian analysis as an uncertainty
and prediction layer.
Which model is best for which question?
Scientific question
Best current model
Evidence status
Publication role
Reason
How does the three-negative rule operate?
Four-State Operational MM
Primary
Main analysis
Directly represents P, N1, N2, C, reset, and recurrence.
Does one common transition matrix miss latent heterogeneity?
Four-State HMM with two hidden regimes
Supported with caution
Secondary analysis
Best AICc and BIC with improved patient-separated prediction.
Should Death probability differ by P, N1, N2, or C?
Shared-Death and Five-State comparison
Mixed
Sensitivity analysis
AICc and LRT favor complexity, whereas BIC and Bayes factors favor restraint.
What is the probability of confirmed C before Death?
Continuous-time competing-risk multi-state model
Supported with caution
Outcome extension
Preserves exposure time and competing first-event outcomes.
Does Bayesian prediction outperform MLE?
No definitive winner
Inconclusive
Methodological adjunct
Log loss improves slightly, but the clustered interval includes no improvement.
Does latent colonization probability decline from P to C?
Positive-conditioned biological HMM
Exploratory
Mechanistic supplement
Strong stage gradient but highly sensitive to initial-state assumptions.
How do negative cultures change near Death or censoring?
GAMM for prediction; Bayesian model for posterior contrasts
Supported with caution / exploratory
Terminal-outcome supplement
Different models answer prediction and posterior-probability questions.
II. The analytical architecture: MM, HMM, Death, and Bayesian layers
One dataset is viewed through four different lenses
①
Observed culture lens
Positive or Negative
Direct laboratory observations without imputing missing specimens.
②
Operational-history lens
P → N1 → N2 → C
Encodes the three-negative protocol and positive resets.
③
Hidden-transition lens
Regime Rt
Allows different transition matrices without changing clinical state labels.
④
Outcome-time lens
Confirmed C or Death
Estimates exposure-time rates, absorption, and horizon probabilities.
Observed culture→Operational evidence history→Latent transition regime→Next state or competing outcome
The models that are most easily confused
Model
Observed state or outcome
Hidden component
Time treatment
Main question
Current role
Four-State Operational MM
P, N1, N2, C
None
Next included culture
What happens next in the three-negative protocol?
Primary
Biological two-state HMM
Positive or negative culture
Clear or Colonized
Observation sequence
What latent biological state may have emitted the culture?
Exploratory
Four-State switching HMM
P, N1, N2, C
One to five transition regimes
Observation sequence
Does the operational transition matrix change over latent regimes?
Secondary predictive model
Four-State plus shared Death
P, N1, N2, C, Death
None
Discrete next transition
Can one common Death probability be used across source states?
Structural baseline
Five-State state-specific Death MM
P, N1, N2, C, Death
None
Discrete next transition
Should Death probability differ by source state?
Sensitivity analysis
Continuous-time competing-risk model
P, N1, N2 transient; C and Death absorbing
None
Exposure days
What is the time-dependent probability of C before Death?
Outcome extension
Bayesian hierarchical terminal model
Observed culture negativity
Patient random effects and posterior uncertainty
Weeks before endpoint
What is the posterior probability of a terminal change?
Exploratory inference
Why “four observed states plus two hidden regimes” is not a six-state clinical model
P Observed
N1 Observed
N2 Observed
C Observed
↑ transition matrix selected by ↑
Hidden regime 1 Lower progression tendency
Hidden regime 2 Higher progression tendency
The Bayesian component is a probabilistic evidence network, not yet a learned causal network
Prior information
Transition regularization
+
Observed transitions
Counts and patient clusters
→
Posterior distribution
Means and credible intervals
↓
Posterior prediction
Held-out log loss and Brier score
Structural evidence
Bayes factor for shared versus state-specific Death
Prior sensitivity
Does the conclusion remain stable?
The Bayesian relationship can be summarized as
\(p(\theta \mid D) \propto p(D \mid \theta)\,p(\theta)\).
The completed analysis uses this logic for transition probabilities, structural Bayes factors,
posterior prediction, and terminal probability contrasts. It does not yet learn a causal graph from the
data, and it should not be described as causal Bayesian-network discovery.
III. Data foundation, denominator flow, and cohort integrity
Four observation denominators belong to four different provenance stages
2,772
Manuscript stated total
→
−50
2,722
Pathogen subtotal
→
−19
2,703
Explicit parsed observations
→
−30
2,673
Prepared observations
Stage
Count
Retained from prior stage
Meaning
Required interpretation
Manuscript stated total
2,772
Reference
Total stated in the manuscript
Must not be silently replaced by a parser count.
Manuscript pathogen subtotal
2,722
98.20%
Arithmetic sum of pathogen counts
The internal 50-observation gap must remain visible.
Explicit parsed observations
2,703
99.30%
Source-derived site-specific positive or negative rows
The 19-observation gap requires source-line provenance.
Prepared observations
2,673
98.89%
Rows retained after deterministic same-day reconciliation
Every one of the 30 reductions should map to a rule.
The analytical cohort narrows in two deliberate steps
Sequence-cohort narrowing. The bars are a cohort funnel rather than a frequency histogram.
321 prepared sequences
98 patients; all patient × pathogen × site chains after reconciliation.
↓ 87 sequences excluded
234 observed-positive Four-State sequences
91 patients; eligible for the primary protocol reconstruction.
↓ 36 short sequences excluded
198 hidden-regime comparison sequences
79 patients; at least three operational observations.
Parser
0 errors
5 information notices
Sequence preparation
8 errors
14 information notices
Reconciliation status
Requires review
Gaps 50, 19, and 30
Outcome linkage
98 / 98
No endpoint-date conflicts
Outcome linkage creates three distinct endpoint classes
17 culture-defined endpoints · audit only in the primary comparator
The overall endpoint linkage is complete, but different analyses use different subsets. The primary
death-versus-censoring terminal analysis uses 44 Death and 37 independent-censoring endpoints. The
Publication Model Evidence Death transition audit links 41 of 44 Death patients under its one-sequence-per-patient
rule. Thirty Death patients had more than one eligible sequence, making the sequence-selection rule an
explicit sensitivity issue rather than an invisible preprocessing detail.
IV. What the primary Four-State Operational MM shows
The protocol state machine makes the principal bottleneck visible
P
Positive
16.78% →
first qualifying negative
N1
One negative
55.64% →
second qualifying negative
N2
Two negatives
73.10% →
third qualifying negative
C
Confirmed clearance
P→P 83.22%
Persistent positivity
N1→P 44.00%
Reset after one negative
N2→P 26.90%
Reset after two negatives
C→P 42.03%
Positive transition after C
Forward progression becomes increasingly favorable after negative evidence accumulates.
The rare N1→N1 too-soon event is omitted from this paired display.
Only 16.78% of next included cultures move from P to N1, making the first qualifying negative the dominant
bottleneck. Once N1 is reached, progression and reset are nearly balanced. After N2, 73.10% confirm C and
26.90% reset to P, showing that the third negative adds meaningful confirmation rather than merely repeating
the second result.
Patient-cluster contrasts show which transitions are statistically convincing
Point estimates and 95% patient-cluster bootstrap intervals. Values above zero favor progression,
maintenance, or persistence according to the contrast label.
Contrast
Difference
95% cluster-bootstrap interval
Holm-adjusted P
Evidence reading
Remain P versus start N1
+66.44 pp
+60.78 to +71.65 pp
0.00600
Strong P bottleneck
N1→N2 versus N1→P
+11.64 pp
+0.35 to +23.08 pp
0.09795
Favorable direction, not familywise significant
N2→C versus N2→P
+46.21 pp
+32.82 to +60.75 pp
0.00600
Strong confirmation evidence
C→C versus C→P
+15.94 pp
−3.90 to +39.40 pp
0.14193
Maintenance and recurrence not clearly separated
The overall monotone forward-progression pattern received 100% patient-cluster bootstrap support.
Nevertheless, the N1 contrast did not survive Holm familywise adjustment. N1 should therefore remain an
unstable evidence-history state rather than being presented as a proven biological turning point.
Immediate progression, eventual clearance, and recurrence form different denominator layers
6.82%
Immediate clean-run product
One uninterrupted P→N1→N2→C path without persistence, reset, or later attempts.
88 / 234
Eventually reached C · 37.61%
Clearance occurred at any later time, including after persistence or reset.
29 / 69
Transition-level C→P · 42.03%
Positive next transition among counted transitions originating from C.
Recurrence denominator map
88 cleared sequences
39 followed cleared sequences
21 recurrent sequences · 53.85% of followed
43 cleared patients
27 followed cleared patients
17 recurrent patients · 62.96% of followed
Endpoint
Estimate
Unit
Follow-up requirement
Correct meaning
Transition-level C→P
29 / 69 = 42.03%
Transition
A counted next transition from C
Positive next transition among observed C-origin transitions
Sequence recurrence among followed
21 / 39 = 53.85%
Sequence
At least one post-clearance transition
At least one C→P event in a followed cleared sequence
Sequence recurrence among all cleared
21 / 88 = 23.86%
Sequence
None
Includes 49 sequences without a counted post-C transition
Patient recurrence among followed
17 / 27 = 62.96%
Patient
At least one followed cleared sequence
At least one recurrent sequence in a followed patient
Time to first recurrence
Median 11 days; IQR 7–31
Time
Observed recurrence
Timing endpoint rather than a recurrence probability
V. HMM evidence: biological states versus hidden transition regimes
The biological HMM shows a strong gradient but weak model robustness
Estimated latent-colonization probability. A logarithmic vertical scale keeps the N2 and C estimates visible.
P · 92.53%
N1 · 25.50%
N2 · 2.96%
C · 0.94%
Interpretation split
The stage gradient is strong, but the model-level evidence remains exploratory because the result is
highly sensitive to the initial hidden-state profile.
BIC penalty relative to the best initial-state profile. Lower is better.
The stationary profile is best by BIC; the positive-conditioned primary profile is approximately 27.08 points worse.
Strong numerical gradient
Every adjacent decline remained supported after clustered bootstrap and Holm adjustment.
Highly sensitive initialization
The preferred profile changes when the initial-state assumption changes.
Exploratory biological interpretation
The gradient does not establish externally validated biological Clear and Colonized states.
The switching-HMM comparison favors two hidden regimes after balancing fit, prediction, and parsimony
Candidate
Parameters
Converged
AICc
BIC
Patient CV log loss
Minimum occupancy
Minimum separation
Four-State MM
6
Yes
2254.700
2288.967
0.506774
100.00%
NE
One hidden regime
6
Yes
2254.700
2288.967
0.506774
100.00%
NE
Two hidden regimes
15
Yes
2164.659
2250.204
0.486243
31.64%
34.13%
Three hidden regimes
26
Yes
2173.920
2321.938
0.483468
27.59%
12.57%
Four hidden regimes
39
Yes
2193.655
2415.218
0.482243
13.15%
10.74%
Five hidden regimes
54
No
2224.257
2530.284
0.483921
5.15%
12.69%
AICc and BIC penalties relative to the best candidate. Both select two hidden regimes.
Patient-separated CV log loss. Four regimes are numerically lowest in one partition, but only slightly.
Occupancy and transition-profile separation deteriorate as the regime count increases.
AICc 2 regimes
BIC 2 regimes
Single CV split 4 regimes
Occupancy 2 regimes
Separation 2 regimes
The four-regime model improves CV log loss over the two-regime model by only about 0.82%, while requiring
39 rather than 15 parameters, increasing BIC by approximately 165 points, reducing minimum occupancy from
31.64% to 13.15%, and reducing minimum transition-profile separation from 34.13% to 10.74%.
Two regimes therefore provide the most defensible compromise.
The two hidden regimes represent transition tendencies, not proven biological subtypes
VI. Does adding Death make the Five-State MM better?
The shared-Death and state-specific Death structures test one narrow modeling question
Four-State plus shared Death
Structural baseline
P
N1
N2
C
↓ one common \(q_D\)
Death
One common probability of Death is imposed across all four operational source states.
Five-State state-specific Death MM
Complexity candidate
P→D
N1→D
N2→D
C→D
↓ four separate probabilities
Absorbing Death state
Three additional degrees of freedom test whether Death probability differs by operational state.
This comparison does not test whether Death is clinically important. It tests whether the available Death
transitions justify separate Death probabilities for P, N1, N2, and C.
The evidence balance is genuinely mixed
Nominal likelihood-ratio test
P = 0.01050
Favors the Five-State candidate
ΔAICc for Five-State
+5.192
Accepts additional fit
Mixed evidence
ΔBIC for Five-State
−11.998
Favors shared-Death restraint
Bayes factor for Five-State
0.06139
Approximately 16:1 against the candidate under the primary prior
Criterion
Result
Direction
What the criterion emphasizes
Likelihood-ratio test
11.239; df 3; P = 0.01050
Five-State
In-sample likelihood gain
ΔAICc
+5.192
Five-State
Predictive-fit emphasis with moderate penalty
ΔBIC
−11.998
Shared Death
Stronger complexity penalty
Exact log Bayes factor
−2.790
Shared Death
Integrated evidence across parameter uncertainty
Equal-prior P(Five-State)
5.78%
Shared Death
Posterior structural probability under the primary prior
Prior sensitivity prevents a definitive Five-State claim
Equal-prior posterior probability of the Five-State structure across prespecified prior scales.
Prior scale 0.5
28.23%
No material structural support
Prior scale 1.0
5.78%
Moderate evidence against Five-State
Prior scale 2.0
0.09%
Strong evidence against Five-State
A structural conclusion that shifts from 28.23% to 0.09% across reasonable prior scales is not robust enough
to justify replacing the shared-Death structure.
Sparse state-specific Death counts explain the disagreement
Forty-one linked Death transitions are distributed very unevenly across the four source states.
Transition
Events
Outgoing transitions
MLE
Posterior mean
95% posterior interval
P→Death
39
1,821
2.14%
2.17%
1.56%–2.91%
N1→Death
1
276
0.36%
0.54%
0.04%–1.70%
N2→Death
0
145
0.00%
0.34%
0.00%–1.75%
C→Death
1
70
1.43%
2.11%
0.15%–6.48%
Sparse Death cells→Higher in-sample flexibility→AICc may improvebutBIC and Bayes factors penalize instability
Practical conclusion: the Five-State Death model should remain a prespecified sensitivity
analysis. It should not replace the Four-State operational model, and the current data do not justify claiming
that Death probability is definitively state-specific.
VII. What MLE and Bayesian analysis actually contributed
MLE and Bayesian estimation solve different parts of the problem
Maximum likelihood estimation
Uses observed transition counts directly
Provides the best-fitting point estimate
Transparent and efficient with adequate cells
Can produce unstable or exact-zero estimates in sparse cells
Bayesian shrinkage avoids interpreting zero observed events as proof that the event is impossible.
Bayesian held-out prediction improves log loss slightly, but not conclusively
Held-out multiclass log loss. Lower is better.
Held-out multiclass Brier score. The two methods are practically indistinguishable.
Metric
Plug-in MLE
Bayesian posterior prediction
Improvement
95% patient-cluster interval
P(improvement)
Decision
Multiclass log loss
0.60805
0.58073
+0.02732
−0.00127 to +0.06357
95.25%
Inconclusive
Multiclass Brier score
0.34516
0.34513
+0.0000268
−0.0000685 to +0.0001184
71.55%
Inconclusive
Useful now
Sparse-cell shrinkage and posterior intervals
Useful now
Prior sensitivity and structural Bayes factors
Not yet established
Consistent patient-separated predictive superiority over MLE
The Bayesian terminal model answers a separate posterior-probability question
Posterior mean differences and 95% credible intervals in percentage points.
Posterior contrast
Mean
95% credible interval
Posterior probability of increase
Interpretation
Death terminal minus earlier
−2.24 pp
−8.19 to +3.58 pp
22.85%
No support for a death-terminal increase
Censoring terminal minus earlier
+9.19 pp
+1.74 to +16.49 pp
99.33%
Strong posterior support for an increase near censoring
Death change minus censoring change
−11.44 pp
−20.99 to −1.76 pp
1.10%
Death-aligned change is probably lower
Bayesian analysis is valuable because it quantifies uncertainty and supports direct probability statements.
Its practical value does not require proving that it always predicts better than MLE. Predictive superiority,
however, remains unproven in the current held-out comparison.
VIII. Competing-risk and terminal-outcome interpretation
The competing-risk model adds time, exposure, and competing absorption
P
⇄
N1
⇄
N2
→
C
or
Death
199 episodes
77 unique patients
54 C-first
Confirmed clearance before Death
83 Death-first
Death before confirmed C
62 right-censored
Neither event observed first
400 / 400 bootstrap
All patient-cluster replicates valid
Transition intensity is estimated as \(q_{ij}=N_{ij}/T_i\), where \(N_{ij}\) is the observed transition count
and \(T_i\) is accumulated exposure time in the source state. This retains the distinction between a
three-day and a fourteen-day interval, although the exact event time remains interval-censored between
cultures.
Accumulated negative evidence increases the fitted probability of C before Death
Eventual model-based first-event probabilities under the fitted homogeneous generator.
Start in P
42.07% C-first
57.93% Death-first
Start in N1
57.81% C-first
42.19% Death-first
Start in N2
77.73% C-first
22.27% Death-first
The increase from P to N1 to N2 is clinically intuitive, but these are fitted first-event probabilities.
They are not validated isolation-release, treatment, or infection-control thresholds.
Horizon maps separate confirmed C, Death, and unresolved transient occupancy
Starting from P
Starting from N1
Starting from N2
Starting state
Horizon
Confirmed C
Death
Still P/N1/N2
Key reading
P
30 days
1.32%
9.05%
89.63%
Most episodes remain unresolved in the near term.
P
90 days
10.59%
23.41%
66.00%
The transient fraction still dominates.
P
365 days
35.67%
51.01%
13.32%
Death exceeds confirmed C at one year.
N1
90 days
34.13%
18.11%
47.76%
Negative evidence materially shifts the near-term balance.
N2
90 days
65.25%
10.04%
24.71%
Most fitted probability has already moved toward confirmed C.
Starting from P, eventual C-before-Death probability is 42.07%, but the probability of already reaching
confirmed C by 90 days is only 10.59%. Reporting only eventual absorption would conceal the 66.00% unresolved
transient fraction at that horizon.
Reverse-time observations and posterior predictions tell a consistent terminal story
Observed patient-weighted negative fractions by week before endpoint.
Bayesian weekly posterior predictions. Death-aligned probability trends downward; censoring-aligned probability trends upward.
Final 4 vs previous 4 weeks
−4.78 pp
One-sided P = 0.8671 · no supported increase
Final 8 vs previous 8 weeks
+3.85 pp
One-sided P = 0.3355 · higher point estimate without support
Death terminal vs earlier
−8.53 pp
95% interval −14.08 to −2.69 pp
Death change point
Not supported
Selected week 3 · bootstrap support 23.60%
Preferred adjusted trend
GAMM
CV log loss 0.623 vs 0.646
P(death-terminal increase)
22.85%
No posterior support for an increase
Patient-window coverage must be read together with the estimated negative fraction
Bars show patient-weighted mean negative fractions. The line shows complete-window coverage among represented patients.
D−1 to D−4
16.38%
19 complete / 34
D−5 to D−8
21.50%
24 complete / 29
D−9 to D−12
16.82%
18 complete / 24
D−13 to D−16
21.06%
13 complete / 18
Missing scheduled weeks remain missing. Later observed panels are never renumbered to compress the timeline,
and an unavailable specimen is never converted to a negative culture.
IX. Integrated evidence map and publication hierarchy
Domain coverage map
Model
Protocol interpretability
Penalized fit
Patient-level prediction
Uncertainty quantification
Death modeling
Biological interpretation
Recommended status
Four-State Operational MM
Strong
Baseline
Baseline
Cluster bootstrap
Not primary purpose
Operational, not biological
Primary model
Four-State HMM · 2 regimes
Moderate
Best AICc and BIC
Improved
Occupancy and separation
Not included
Transition regimes only
Best secondary extension
Five-State state-specific Death MM
Moderate
AICc favorable; BIC unfavorable
Not decisive
Posterior intervals and Bayes factor
Direct
No biological subtype
Sensitivity model
Bayesian Five-State prediction
Moderate
Same structure
Slight improvement; inconclusive
Strong
Direct
Not causal
Uncertainty adjunct
Competing-risk multi-state model
Strong outcome interpretation
Different likelihood target
Model-based horizons
Cluster bootstrap
Direct and time-aware
First-event interpretation
Outcome extension
Biological Clear/Colonized HMM
Indirect
Relative fit favorable
Absolute adequacy uncertain
Posterior probabilities
Not primary purpose
Highly assumption-sensitive
Exploratory
Terminal GAMM / Bayesian model
Not a clearance-rule model
Criteria disagree
GAMM preferred by CV
Posterior contrasts
Endpoint-relative
Observational only
Exploratory outcome analysis
Qualitative two-dimensional evidence map
Lower predictive support
Moderate predictive support
Higher predictive support
Higher interpretability
Shared-Death structural baseline
Competing-risk model
Four-State Operational MM
Moderate interpretability
Five-State Death sensitivity
Bayesian transition prediction
Two-regime Four-State HMM
Lower interpretability
Biological HMM under unstable initialization
Bayesian terminal model
Terminal GAMM
This map is an interpretive synthesis rather than a new statistical analysis. It positions each model by the
clarity of its clinical meaning and the strength of its current predictive evidence.
Secondary evidence that one common transition matrix does not fully describe the operational process.
Main result 3
P/N1/N2/C/Death competing-risk model
Exposure-time transition rates, C-before-Death absorption, and horizon probabilities.
Sensitivity analysis
Shared versus state-specific Death
Mixed evidence should be reported without a universal winner claim.
Methodological adjunct
MLE versus Bayesian prediction
Bayesian shrinkage is useful, but predictive superiority remains inconclusive.
Exploratory supplement
Biological HMM and terminal models
Directional and predictive findings with substantial assumption and identifiability limits.
The observed surveillance process is most transparently represented by the protocol-aligned Four-State
Operational MM. A two-hidden-regime switching extension improves penalized fit and patient-separated
prediction, supporting latent transition heterogeneity while preserving P, N1, N2, and C as the observed
states. Evidence that Death requires state-specific discrete transition probabilities is mixed, and
Bayesian posterior prediction does not yet show conclusive held-out superiority over plug-in MLE.
X. Claims supported by the current results and claims that should be avoided
Claim boundary map
Supported or defensible
The Four-State Operational MM is the primary protocol-aligned model.
P→N1 is the dominant operational bottleneck.
N2→C is substantially more likely than N2→P.
Forward progression becomes more favorable as negative evidence accumulates.
A two-hidden-regime HMM is supported as a secondary transition model.
The two-regime HMM improves penalized fit and patient-separated prediction.
C-before-Death probability rises from P to N1 to N2.
The final-four-week negative-surge hypothesis is not supported.
The final-eight-week point estimate is higher but unconfirmed.
Recurrence depends materially on unit and follow-up denominator.
The Five-State MM is definitively better than the shared-Death baseline.
Bayesian posterior prediction is conclusively superior to MLE.
The two hidden regimes are validated biological subtypes.
The biological HMM has identified true Clear and Colonized states.
Terminal illness caused the observed culture pattern.
Confirmed local C is a validated release or treatment threshold.
HMM emission sensitivity and specificity equal laboratory test accuracy.
C→P equals patient-level recurrence incidence.
A missing or unobtainable specimen can be treated as negative.
XI. Highest-priority work before final publication
Publication-readiness roadmap
Before submission
Resolve data provenance
Explain the 50-observation manuscript arithmetic gap.
Trace the 19-observation manuscript-to-parser difference.
Trace all 30 parser-to-preparation reductions.
Review the eight sequence-preparation errors individually.
Model-stability phase
Repeat internal validation
Repeat patient-level CV over multiple fold assignments and seeds.
Report the distribution of selected hidden-regime counts.
Test alternative one-sequence-per-Death linkage rules.
Assess the influence of long Death-linkage gaps.
Future validation
Strengthen generalizability
Develop a hierarchical Bayesian transition model with partial pooling.
Compare homogeneous, semi-Markov, and interval-censored formulations.
Perform temporal validation in a later cohort.
Perform external validation in another institution.
Priority matrix
Work item
Scientific impact
Required before main submission?
Primary risk addressed
Expected effect on the conclusion
Observation-level provenance audit
Very high
Yes
Data credibility
Determines whether all later modeling is defensible.
Repeated patient-level CV
Very high
Strongly recommended
Hidden-regime order instability
Could strengthen or weaken the two-regime recommendation.
Alternative Death linkage rules
High
Recommended
One-sequence-per-patient selection bias
Tests robustness of the Five-State conclusion.
Hierarchical Bayesian partial pooling
High
No
Sparse state-specific Death cells
May improve calibration and stabilize state-specific effects.
External validation
Very high
No
Generalizability
Determines whether hidden regimes and outcome probabilities reproduce.
Formal Bayesian network
Potentially high
No
Unspecified causal structure
Should follow node, temporal-order, and causal-purpose prespecification.
XII. Final integrated answer
Final ranking and role assignment
Rank
Model or analysis
Primary strength
Primary limitation
Final role
1
Four-State Operational MM
Protocol alignment and transparent interpretation
One common observed-state transition structure
Primary manuscript model
2
Four-State HMM with two hidden regimes
Penalized fit and latent transition heterogeneity
Regime count still needs repeated CV
Best secondary model
3
P/N1/N2/C/Death competing-risk model
Time-aware C-before-Death inference
Homogeneous continuous-time approximation
Outcome extension
4
Five-State state-specific Death MM
Direct test of state-specific Death probabilities
Sparse events and mixed evidence
Sensitivity analysis
5
Bayesian transition prediction
Shrinkage and uncertainty
Predictive superiority is inconclusive
Methodological adjunct
6
Biological HMM and terminal models
Mechanistic and temporal supporting information
Initialization, identifiability, and observational limits
Exploratory supplement
Explain the protocol
Four-State MM
Capture hidden heterogeneity
Two-regime HMM
Model C versus Death over time
Competing risks
Final answer: the evidence does not support replacing the Four-State MM with one universal
alternative. The strongest publication structure is a layered hierarchy: the Four-State MM as the primary
protocol model, the two-hidden-regime Four-State HMM as the supported predictive extension, and the
competing-risk model as the Death-aware outcome extension. The Five-State Death structure and Bayesian
predictive advantage should be reported transparently as unresolved or sensitivity findings.
nGeneMDRO complete analysis 해석: MM, HMM, Death, MLE, Bayesian 모델을 위한 시각적 근거 지도
Canonical report
36개 section
Complete result package
Primary model
4상태 MM
프로토콜 설명
Best secondary model
2-regime HMM
전이 이질성
Death structure
Mixed evidence
5상태 우월성 미확립
Bayesian prediction
Inconclusive
유용한 불확실성 계층
Overall evidence
Mixed
질문별로 다른 답
핵심 결론은 한 모델이 모든 비교에서 승리했다는 것이 아닙니다.
가장 방어적인 결론은 layered model hierarchy입니다. 프로토콜 설명에는 4상태 운영적 MM,
추가적인 예측구조에는 2개 숨은 체제의 4상태 HMM, Death-aware outcome probability에는
연속시간 경쟁위험 모델이 각각 우선됩니다.
Structural evidence
공통 Death와 상태별 Death의 Bayes factor
Prior sensitivity
결론이 prior에 따라 흔들리는가?
Bayesian 관계는 \(p(\theta \mid D) \propto p(D \mid \theta)\,p(\theta)\)로 요약할 수 있습니다.
현재 분석은 이 구조를 전이확률, structural Bayes factor, posterior prediction,
terminal probability contrast에 사용합니다. 자료에서 causal graph를 별도로 학습한 것은 아니므로
causal Bayesian-network discovery로 표현해서는 안 됩니다.
III. 자료 기반, 분모 흐름, cohort 정합성
네 관찰 분모는 서로 다른 provenance 단계입니다
2,772
논문 기재 총계
→
−50
2,722
병원체별 소계
→
−19
2,703
명시적으로 파싱된 관찰
→
−30
2,673
준비된 관찰
단계
수
이전 단계 대비 유지율
의미
필수 해석
논문 기재 총계
2,772
기준
논문에 기재된 전체 수
Parser count로 조용히 대체해서는 안 됩니다.
논문 병원체 소계
2,722
98.20%
병원체별 수의 산술 합계
내부 50건 차이를 표시해야 합니다.
명시적 파싱 관찰
2,703
99.30%
검체부위별 positive 또는 negative row
19건 차이에 source-line provenance가 필요합니다.
준비된 관찰
2,673
98.89%
Same-day reconciliation 후 유지된 row
30건 감소가 모두 결정론적 규칙과 연결되어야 합니다.
분석 cohort는 의도적으로 두 단계에서 좁아집니다
Sequence cohort funnel입니다. 빈도 histogram이 아니라 분석대상의 단계적 축소를 표시합니다.
준비된 sequence 321개
98명, reconciliation 후 모든 patient × pathogen × site chain.
전체 endpoint linkage는 완전하지만 분석별 subset은 다릅니다. Primary death-versus-censoring terminal
analysis는 Death 44명과 independent censoring 37명을 사용합니다. Publication Model Evidence의
Death transition audit는 환자당 하나의 sequence 규칙에 따라 44명 중 41명을 연결합니다.
Death 환자 30명은 eligible sequence가 여러 개였으므로 sequence 선택규칙은 숨겨진 preprocessing이
아니라 명시적인 sensitivity issue입니다.
IV. 주 분석인 4상태 운영적 MM이 보여주는 것
프로토콜 state machine은 가장 큰 bottleneck을 명확히 보여줍니다
P
Positive
16.78% →
첫 qualifying negative
N1
음성 1회
55.64% →
두 번째 qualifying negative
N2
음성 2회
73.10% →
세 번째 qualifying negative
C
확인된 clearance
P→P 83.22%
Positive persistence
N1→P 44.00%
음성 1회 후 reset
N2→P 26.90%
음성 2회 후 reset
C→P 42.03%
C 이후 positive transition
음성 근거가 누적될수록 forward progression이 더 유리해집니다.
드문 N1→N1 too-soon event는 이 paired display에서 제외했습니다.
다음 포함 배양에서 P에서 N1로 이동하는 비율은 16.78%에 불과하여 첫 qualifying negative가 가장 큰
bottleneck입니다. N1에 도달한 뒤에는 진행과 reset이 거의 균형을 이룹니다. N2 이후에는 73.10%가
C를 확인하고 26.90%가 P로 reset되므로 세 번째 음성은 두 번째 결과의 단순 반복이 아니라 의미 있는
confirmation step입니다.
Patient-cluster contrast는 어떤 전이가 통계적으로 설득력 있는지 보여줍니다
Point estimate와 95% patient-cluster bootstrap interval입니다.
0보다 큰 값은 contrast label에 따라 progression, maintenance 또는 persistence를 지지합니다.
Contrast
차이
95% cluster-bootstrap interval
Holm-adjusted P
근거 해석
P 유지 대 N1 시작
+66.44 pp
+60.78 to +71.65 pp
0.00600
강한 P bottleneck
N1→N2 대 N1→P
+11.64 pp
+0.35 to +23.08 pp
0.09795
방향은 유리하지만 familywise significance 없음
N2→C 대 N2→P
+46.21 pp
+32.82 to +60.75 pp
0.00600
강한 confirmation evidence
C→C 대 C→P
+15.94 pp
−3.90 to +39.40 pp
0.14193
유지와 recurrence가 명확히 분리되지 않음
전체적인 monotone forward-progression pattern은 100% patient-cluster bootstrap support를 받았습니다.
그러나 N1 contrast는 Holm familywise adjustment를 통과하지 못했습니다. 따라서 N1은 입증된
biological turning point가 아니라 불안정한 evidence-history state로 유지하는 것이 적절합니다.
Immediate progression, eventual clearance, recurrence는 서로 다른 denominator layer입니다
6.82%
Immediate clean-run product
Persistence, reset, later attempt 없이 한 번에 P→N1→N2→C로 진행한 경로입니다.
88 / 234
Eventually reached C · 37.61%
Persistence 또는 reset 후 다시 시도한 경우까지 포함한 eventual clearance입니다.
29 / 69
Transition-level C→P · 42.03%
C에서 출발한 counted transition 중 다음 transition이 positive였던 비율입니다.
Recurrence denominator map
Cleared sequence 88개
Follow-up이 있는 cleared sequence 39개
Recurrent sequence 21개 · followed의 53.85%
Cleared patient 43명
Follow-up이 있는 cleared patient 27명
Recurrence patient 17명 · followed의 62.96%
Endpoint
추정치
분석단위
Follow-up 조건
올바른 의미
Transition-level C→P
29 / 69 = 42.03%
Transition
C에서 출발한 다음 transition 존재
관찰된 C-origin transition 중 positive next transition
해석을 둘로 나누어야 합니다
Stage gradient는 강하지만 initial hidden-state profile에 매우 민감하므로 model-level evidence는
exploratory로 남습니다.
최적 initial-state profile 대비 BIC penalty입니다. 낮을수록 좋습니다.
Stationary profile이 BIC상 최적이며 positive-conditioned primary profile은 약 27.08 points 불리합니다.
강한 numerical gradient
모든 인접 감소가 clustered bootstrap과 Holm adjustment 이후에도 지지됩니다.
Highly sensitive initialization
Initial-state assumption에 따라 최적 profile이 달라집니다.
Exploratory biological interpretation
실제 Clear와 Colonized 상태가 외부 검증된 것은 아닙니다.
Switching-HMM 비교는 적합도, 예측, 단순성을 함께 볼 때 2개 체제를 지지합니다
후보
Parameter
Converged
AICc
BIC
Patient CV log loss
최소 occupancy
최소 separation
4상태 MM
6
Yes
2254.700
2288.967
0.506774
100.00%
NE
숨은 체제 1개
6
Yes
2254.700
2288.967
0.506774
100.00%
NE
숨은 체제 2개
15
Yes
2164.659
2250.204
0.486243
31.64%
34.13%
숨은 체제 3개
26
Yes
2173.920
2321.938
0.483468
27.59%
12.57%
숨은 체제 4개
39
Yes
2193.655
2415.218
0.482243
13.15%
10.74%
숨은 체제 5개
54
No
2224.257
2530.284
0.483921
5.15%
12.69%
최적 후보 대비 AICc와 BIC penalty입니다. 두 기준 모두 2개 체제를 선택합니다.
Patient-separated CV log loss입니다. 한 partition에서는 4개 체제가 가장 낮지만 차이는 작습니다.
Regime 수가 증가할수록 occupancy와 transition-profile separation이 악화됩니다.
AICc 2개 체제
BIC 2개 체제
Single CV split 4개 체제
Occupancy 2개 체제
Separation 2개 체제
4개 체제 모델은 2개 체제 모델보다 CV log loss를 약 0.82% 개선하지만, parameter는 15개에서 39개로
증가하고 BIC는 약 165 points 악화됩니다. 최소 occupancy는 31.64%에서 13.15%로,
최소 transition-profile separation은 34.13%에서 10.74%로 감소합니다.
따라서 2개 체제가 가장 방어적인 균형입니다.
두 숨은 체제는 transition tendency이며 입증된 biological subtype이 아닙니다
Hidden regime 1Occupancy 68.36%
Progression score
−0.116
P persistence 92.45%
N2→C 42.07%
C→P 83.54%
허용 표현: 낮은 clearance-progression transition regime
Hidden regime 2Occupancy 31.64%
Progression score
+0.514
P→N1 54.34%
N2→C 79.30%
C maintenance 61.45%
허용 표현: 높은 clearance-progression transition regime
방어 가능한 표현
Latent transition phenotype
Lower- and higher-progression regime
Transition-matrix heterogeneity
피해야 할 표현
Biological subtype
Immune phenotype
Treatment-response class
Validated patient subgroup
VI. Death를 추가한 5상태 MM이 더 좋은가
공통 Death와 상태별 Death 구조는 좁고 명확한 한 질문을 검정합니다
4상태 + 공통 Death
Structural baseline
P
N1
N2
C
↓ 하나의 공통 \(q_D\)
Death
네 운영상태 모두에 하나의 공통 Death probability를 적용합니다.
상태별 Death 5상태 MM
Complexity candidate
P→D
N1→D
N2→D
C→D
↓ 네 개의 별도 probability
Absorbing Death state
세 개의 추가 degree of freedom으로 운영상태별 Death probability 차이를 검정합니다.
이 비교는 Death가 임상적으로 중요한지를 검정하는 것이 아닙니다. 현재 Death transition 수가
P, N1, N2, C별 별도 probability를 정당화하는지를 검정합니다.
근거의 저울은 실제로 양쪽으로 갈립니다
Nominal likelihood-ratio test
P = 0.01050
5상태 후보 지지
Five-State ΔAICc
+5.192
추가 적합도를 수용
Mixed evidence
Five-State ΔBIC
−11.998
공통 Death 단순성 지지
Five-State Bayes factor
0.06139
Primary prior에서 후보에 약 16:1 불리
합리적인 prior 범위에서 structural probability가 28.23%에서 0.09%까지 변하므로 공통 Death 구조를
5상태 구조로 대체할 만큼 견고한 결론으로 볼 수 없습니다.
상태별 Death count의 희소성이 기준 불일치를 설명합니다
연결된 Death transition 41개가 네 출발상태에 매우 불균형하게 분포합니다.
Transition
Event
Outgoing transition
MLE
Posterior mean
95% posterior interval
P→Death
39
1,821
2.14%
2.17%
1.56%–2.91%
N1→Death
1
276
0.36%
0.54%
0.04%–1.70%
N2→Death
0
145
0.00%
0.34%
0.00%–1.75%
C→Death
1
70
1.43%
2.11%
0.15%–6.48%
Sparse Death cell→높은 in-sample flexibility→AICc 개선 가능그러나BIC와 Bayes factor는 불안정성을 벌점화
실무적 결론: 5상태 Death model은 prespecified sensitivity analysis로 유지하는 것이
적절합니다. 4상태 운영적 모델을 대체해서는 안 되며 현재 자료는 Death probability가 확실히
state-specific이라고 주장할 근거를 제공하지 않습니다.
Bayesian held-out prediction은 log loss를 조금 개선하지만 결론은 불확실합니다
Held-out multiclass log loss입니다. 낮을수록 좋습니다.
Held-out multiclass Brier score입니다. 두 방법은 실질적으로 거의 같습니다.
Metric
Plug-in MLE
Bayesian posterior prediction
Improvement
95% patient-cluster interval
P(improvement)
결론
Multiclass log loss
0.60805
0.58073
+0.02732
−0.00127 to +0.06357
95.25%
Inconclusive
Multiclass Brier score
0.34516
0.34513
+0.0000268
−0.0000685 to +0.0001184
71.55%
Inconclusive
현재 이미 유용함
Sparse-cell shrinkage와 posterior interval
현재 이미 유용함
Prior sensitivity와 structural Bayes factor
아직 확립되지 않음
MLE보다 일관된 patient-separated predictive superiority
Bayesian terminal model은 별도의 posterior-probability 질문에 답합니다
Percentage point 단위의 posterior mean difference와 95% credible interval입니다.
Posterior contrast
Mean
95% credible interval
증가 posterior probability
해석
Death terminal minus earlier
−2.24 pp
−8.19 to +3.58 pp
22.85%
Death-terminal increase 지지 없음
Censoring terminal minus earlier
+9.19 pp
+1.74 to +16.49 pp
99.33%
Censoring 근처 증가에 대한 강한 posterior support
Death change minus censoring change
−11.44 pp
−20.99 to −1.76 pp
1.10%
Death-aligned change가 더 낮을 가능성이 큼
Bayesian 분석은 uncertainty를 정량화하고 직접적인 probability statement를 제공한다는 점에서
유용합니다. 항상 MLE보다 더 잘 예측해야만 가치가 있는 것은 아닙니다. 그러나 현재 자료에서는
predictive superiority가 입증되지 않았습니다.
VIII. 경쟁위험 및 terminal-outcome 해석
경쟁위험 모델은 시간, exposure, competing absorption을 추가합니다
P
⇄
N1
⇄
N2
→
C
또는
Death
Episode 199개
Unique patient 77명
C-first 54개
Death 이전에 confirmed clearance
Death-first 83개
Confirmed C 이전에 Death
Right-censored 62개
두 event 중 어느 것도 먼저 관찰되지 않음
Bootstrap 400 / 400
모든 patient-cluster replicate 유효
Transition intensity는 \(q_{ij}=N_{ij}/T_i\)로 추정됩니다. \(N_{ij}\)는 관찰된 transition count,
\(T_i\)는 source state에서 누적된 exposure time입니다. 따라서 3일 interval과 14일 interval이
같은 time at risk로 계산되지 않습니다. 다만 정확한 event time은 배양 사이에서 interval-censored입니다.
음성 근거가 누적될수록 fitted C-before-Death probability가 증가합니다
P에서 N1, N2로 갈수록 C-first probability가 높아지는 것은 임상적으로 직관적입니다.
그러나 이 값은 fitted first-event probability이며 검증된 격리 해제, 치료, 감염관리 threshold가 아닙니다.
Horizon map은 confirmed C, Death, unresolved transient occupancy를 동시에 보여줍니다
P에서 시작
N1에서 시작
N2에서 시작
시작상태
Horizon
Confirmed C
Death
Still P/N1/N2
핵심 해석
P
30일
1.32%
9.05%
89.63%
단기에는 대부분 unresolved입니다.
P
90일
10.59%
23.41%
66.00%
Transient fraction이 여전히 가장 큽니다.
P
365일
35.67%
51.01%
13.32%
1년에는 Death가 confirmed C보다 높습니다.
N1
90일
34.13%
18.11%
47.76%
음성 근거가 단기 balance를 크게 바꿉니다.
N2
90일
65.25%
10.04%
24.71%
대부분의 fitted probability가 confirmed C 방향으로 이동합니다.
P에서 시작한 eventual C-before-Death probability는 42.07%이지만 90일까지 이미 confirmed C에 도달한
probability는 10.59%입니다. Eventual absorption만 보고하면 같은 시점에 남아 있는 66.00%의 unresolved
transient fraction이 가려집니다.
Directional 또는 predictive finding은 있으나 assumption과 identifiability limit가 큽니다.
관찰된 감시과정은 프로토콜과 직접 일치하는 4상태 운영적 MM으로 가장 투명하게 표현됩니다.
두 개의 숨은 전이 체제를 가진 switching extension은 P, N1, N2, C를 관찰상태로 유지하면서
penalized fit과 patient-separated prediction을 개선합니다. 반면 Death가 운영상태별로 서로 다른
discrete transition probability를 가져야 한다는 근거는 혼재되어 있으며 Bayesian posterior
prediction이 MLE보다 확실히 우월하다는 근거도 충분하지 않습니다.
최종 답: 현재 근거는 4상태 MM을 하나의 보편적 대안으로 대체하는 결론을 지지하지
않습니다. 가장 강한 논문 구조는 layered hierarchy입니다. 4상태 MM을 주 protocol model로,
2개 숨은 체제의 4상태 HMM을 지지되는 predictive extension으로, 경쟁위험 모델을 Death-aware
outcome extension으로 배치하는 것이 적절합니다. Five-State Death structure와 Bayesian predictive
advantage는 해결되지 않은 sensitivity finding으로 투명하게 보고해야 합니다.
Written on August 5, 2026
Clinical meaning hidden in the longitudinal MDRO dataset: what nGeneMDRO adds beyond the original study (Written August 5, 2026)
nGeneMDRO is most appropriately framed as a derived process-dynamics study. The original manuscript established that clearance differed by anatomic reservoir and pathogen, that mortality was associated more with host and site than pathogen identity, and that co-colonization was strongly asymmetric around CRE. The derived analysis asks a different question: what does the culture history available today imply about the episode’s next path?
The central clinical interpretation is that MDRO clearance in this long-term-care facility cohort was not a binary event. It was difficult to initiate, became progressively more credible as qualifying negative cultures accumulated, often required repeated attempts, could become positive again soon after operational clearance, and frequently competed with death or prolonged unresolved carriage.
I. The original and derived studies describe two different axes
Axis
Original manuscript
nGeneMDRO-derived analysis
Clinical meaning
Vertical hierarchy
Local site, all sites for one pathogen, and whole-patient clearance
Preserves one patient × one pathogen × one site as the local analytical unit
Local C must not be substituted for pathogen-level clearance or patient-level clearance
Horizontal trajectory
Time to a retrospectively confirmed clearance endpoint
P → N1 → N2 → C, positive resets, post-C re-positivity, censoring, and Death
The accumulated sequence contains information that a single positive/negative label discards
A critical interpretive boundary follows from this map. C in nGeneMDRO is confirmed local clearance for one patient–pathogen–site sequence. It is not automatically pathogen-level clearance, patient-level clearance, or authorization to discontinue all transmission-based precautions.
II. The first negative is the main bottleneck, and the third negative remains meaningful
Beginning a negative run was difficult
While an episode was in P, only 16.78% of next included cultures moved to N1; 83.22% remained in P. The principal barrier was therefore leaving persistent positivity, not merely completing the final confirmatory culture.
This finding suggests that future interventions should first be evaluated for whether they help an episode leave P. Time to the first qualifying negative may be a sensitive intermediate endpoint, provided that it is not mislabeled as confirmed clearance.
One negative was fragile, whereas two negatives were substantially stronger
From N1, 55.64% progressed to N2 and 44.00% reset to P. This remained a near-balanced state after multiplicity was considered.
From N2, 73.10% reached C and 26.90% reset to P, a strongly supported difference. The third negative therefore retained non-redundant confirmatory value.
Clearance onset and confirmation are different clinical times
The original time-to-clearance endpoint is dated to the first negative of a subsequently successful three-negative run. That is a retrospective onset date. At the time of N1, the future second and third negatives are unknown.
The four-state reconstruction prevents future information from leaking backward into real-time prediction and avoids calling a first negative “clearance” before the confirming sequence exists.
Clearance often required more than one attempt
The uninterrupted P → N1 → N2 → C product was only 6.82%, whereas 37.61% of eligible sequences eventually reached C. Persistence, failed runs, resets, and later attempts were therefore integral to the observed clearance process.
P
Positive
16.78%
→
N1
First negative
55.64%
→
N2
Two negatives
73.10%
→
C
Local confirmation
P remains P: 83.22% · N1 resets to P: 44.00% · N2 resets to P: 26.90% · observed C → P transition: 42.03%
III. Operational clearance was frequently followed by detected re-positivity
C did not mean permanent biological eradication. The recurrence estimate changed substantially according to the analytical unit and whether post-clearance surveillance continued.
Endpoint
Estimate
Correct clinical wording
C → P transition
29/69 = 42.03%
Positive result among counted next transitions observed from C
Followed cleared sequences
21/39 = 53.85%
At least one detected re-positivity among sequences with post-clearance follow-up
All cleared sequences
21/88 = 23.86%
Detected re-positivity when sequences without follow-up remain in the denominator
Followed cleared patients
17/27 = 62.96%
At least one recurrent sequence among patients with post-clearance follow-up
All cleared patients
17/43 = 39.53%
Detected recurrence among all patients with at least one local C
The median observed time to first re-positivity was 11 days, with an interquartile range of 7–31 days. This makes the early post-clearance period clinically important, but it does not distinguish persistent colonization below detection, intermittent shedding, sampling variation, laboratory variation, or true reacquisition. The most defensible term is detected post-clearance re-positivity.
Targeted early post-clearance retesting is a reasonable hypothesis for prospective evaluation. The current results do not establish a mandatory new surveillance schedule.
IV. Current state provides dynamic information about clearance before Death
In the continuous-time competing-risk model, the estimated probability of confirmed local C occurring before Death increased as negative evidence accumulated.
Starting state
C before Death
Death before C
Clinical reading
P
42.07%
57.93%
The episode remains distant from confirmed local clearance
N1
57.81%
42.19%
The balance improves, but reset remains common
N2
77.73%
22.27%
Confirmed C is substantially more likely under the fitted model
Starting from P at 90 days, the fitted probabilities were 10.59% confirmed C, 23.41% Death, and 66.00% still in P/N1/N2. The dominant short-term clinical result was therefore prolonged unresolved surveillance status.
These are episode-based model estimates, not bedside release thresholds. They also do not show that N1 or N2 prevents death. N2 is structurally closer to C, and the separate evidence for state-specific death hazards was mixed.
V. No terminal surge in negative cultures was demonstrated
The prespecified final-four-week hypothesis was negative
Among 16 complete paired patients, final four minus previous four weeks was −4.78 percentage points, with a 95% interval of −12.60 to +3.81 and a one-sided P value of 0.8671.
The death-aligned terminal window was lower than the earlier window
The paired terminal-minus-earlier difference was −8.53 percentage points, with a 95% interval of −14.08 to −2.69.
Death-aligned and independent-censoring trajectories diverged
In the adjusted Bayesian model, death-aligned negativity changed from 22.62% to 20.38%, whereas independent-censoring-aligned negativity changed from 24.06% to 33.25%. The probability that the death-aligned change exceeded the censoring-aligned change was only 1.10%.
Missing or unobtainable specimens must not be converted into negative cultures. The present analysis does not support spontaneous terminal decolonization and does not establish that persistent positivity causes death.
VI. The data contain hidden transition heterogeneity, but not yet validated biological subtypes
A binary positive/negative model was inadequate
Pathogen-by-site transition homogeneity and first-order memorylessness were strongly violated. Although the binary HMM fit relatively better than the binary Markov model, neither model reproduced all observed patterns. The preceding sequence therefore matters.
A two-regime switching HMM improved fit and patient-separated prediction
Transition
Lower-progression regime
Higher-progression regime
Interpretation
P → N1
7.55%
54.34%
The same P state can conceal very different chances of beginning a negative run
N1 → N2
28.51%
69.77%
A first negative may be unstable in one context and strongly progressive in another
N2 → C
42.07%
79.30%
The surrounding transition context remains important after two negatives
C → C
16.46%
61.45%
Post-C durability also differs substantially between regimes
These regimes may reflect unmeasured antibiotic exposure, devices, source control, respiratory instrumentation, wound burden, microbiome disruption, organism burden, or other time-varying factors. They are mathematical transition patterns, not “two patient types.”
The biological HMM supports the ordering of evidence states, not a true individual probability
Mean latent-colonization probability declined from 92.53% in P to 25.50% in N1, 2.96% in N2, and 0.94% in C. This is coherent with accumulating negative evidence.
However, the model was highly sensitive to the initial-state assumption, and its sensitivity and specificity are model emissions without an external microbiological gold standard. These estimates must not be presented as validated laboratory test characteristics or individual biological truth.
VII. What can be said clinically—and what should not be claimed
Defensible now
Hypothesis-generating
Not supported by the current data
The first qualifying negative is the main bottleneck
Interventions may be evaluated by their ability to move episodes from P to N1
A first negative is equivalent to clearance
N1 is fragile; N2 is substantially more favorable; the third negative remains confirmatory
Current state may support dynamic counseling or risk-stratified follow-up after validation
N2 causally reduces mortality
Detected post-clearance re-positivity is frequent among followed units and often early
Targeted early post-clearance retesting merits prospective evaluation
A universal new retesting schedule is already justified
No final-four-week rise in observed negativity was demonstrated
Joint modeling of specimen availability and culture outcome may clarify terminal patterns
Persistent positivity predicts or causes imminent death
Two latent transition regimes improved the complementary model
Clinical covariates may explain regime membership or switching
The regimes are validated biological phenotypes
Local C is an operational patient–pathogen–site endpoint
Durable 30-, 60-, and 90-day local clearance should be studied
Local C equals permanent eradication, PLC, PtLC, or automatic de-isolation
VIII. Data boundaries and the strongest publication framing
Issue
Why it matters
2,772 → 2,722 → 2,703 → 2,673 observations
The manuscript total, pathogen subtotal, parsed observations, and reconciled observations represent different stages. The initial 50-observation manuscript arithmetic gap and eight error-level preparation issues require source-level audit.
Different analytical units
Observations, transitions, local sequences, patients, and death-linked episodes are not interchangeable. Numerators, denominators, unique-patient counts, and clustering must remain visible.
Selective post-clearance follow-up
Recurrence proportions are detection-dependent. A recurrent-event or multi-state time-to-event model is needed for durable clearance.
Informative observation process
Sampling intensity was associated with observed negativity. More testing must not be interpreted as causing clearance.
Co-colonization remains partly unused
The original CRE-centered asymmetric network supports a future coupled multi-pathogen state model rather than treating every pathogen–site episode as independent.
Publication-safe synthesis: Within this prospective LTCF cohort, MDRO clearance behaved as a history-dependent multistep process. The principal barrier was initiation of a qualifying negative run. Accumulating negative evidence progressively favored confirmed local clearance, but intermediate states could reset and operational clearance could be followed by detected re-positivity. Death competed with clearance, most episodes starting from P remained unresolved at 90 days, and no increase in culture negativity was demonstrated near death. These findings support explicit interpretation of serial evidence, resets, follow-up, and competing outcomes, while not equating local clearance with permanent eradication, patient-level clearance, or a causal reduction in mortality.
The strongest identity for the second study is therefore dynamic surveillance and clearance-pathway analysis, not validation of a de-isolation rule. The most valuable hidden information is that the sequence itself carries clinical meaning.
장기 MDRO 데이터에 숨어 있던 임상적 의미: 원 논문을 넘어 nGeneMDRO가 추가로 보여주는 것
nGeneMDRO는 원 논문의 분석을 반복하는 도구라기보다 파생 과정동역학 연구로 보는 것이 가장 적절합니다. 원 논문이 해부학적 저장소와 병원체에 따른 제거 차이, 병원체 종류보다 숙주와 부위에 더 연관된 사망, CRE 중심의 비대칭 동시 집락화를 보여주었다면, 파생 분석은 다른 질문을 다룹니다. 즉, 현재까지 확보된 배양 이력이 해당 에피소드의 다음 경로에 관하여 무엇을 말해주는가입니다.
핵심 임상 해석은 다음과 같습니다. 이 장기요양병원 코호트에서 MDRO 제거는 이분형 사건이 아니었습니다. 제거는 시작하기 어려웠고, 적격 음성이 누적될수록 점차 신뢰도가 높아졌으며, 여러 차례의 시도가 필요한 경우가 많았습니다. 조작적 제거 뒤에도 조기 재양성이 가능했고, 제거가 완성되기 전에 사망하거나 장기간 미해결 상태로 남는 경우가 흔했습니다.
I. 원 연구와 파생 연구는 서로 다른 두 축을 설명합니다
축
원 논문
nGeneMDRO 파생 분석
임상적 의미
수직 계층
국소 부위, 한 병원체의 모든 부위, 환자 전체의 제거
한 환자 × 한 병원체 × 한 부위를 국소 분석단위로 유지
국소 C를 병원체 수준 제거 또는 환자 수준 제거로 대체해서는 안 됩니다
수평 궤적
회고적으로 확인된 제거 종결점까지의 시간
P → N1 → N2 → C, 양성 재설정, C 후 재양성, 중도절단, 사망
연속 이력은 양성·음성 한 값이 버리는 정보를 보존합니다
숨은 구조
부위, 병원체, 숙주요인, 동시 집락화
전이 병목, 잠재 전이 체제, 생물학적 HMM, 말기 궤적, 경쟁위험
현재 결과가 같아도 향후 경로는 크게 다를 수 있습니다
집계
부위 → 병원체 → 환자
시간
P → N1 → N2 → C ↔ P
통합 임상 상태
저장소 + 병원체 + 누적 근거 + 경쟁 결과
이 구조에서 반드시 지켜야 할 경계가 있습니다. nGeneMDRO의 C는 한 환자–한 병원체–한 부위에서 확인된 국소 제거입니다. 이는 자동으로 병원체 수준 제거, 환자 수준 제거, 또는 모든 전파주의 해제를 뜻하지 않습니다.
II. 첫 음성이 가장 큰 병목이며, 세 번째 음성도 여전히 의미가 있습니다
음성 연속과정을 시작하는 것이 어려웠습니다
P 상태에서 다음 포함 배양 중 N1으로 진행한 비율은 16.78%뿐이었고, 83.22%는 P에 남았습니다. 가장 큰 장벽은 마지막 확인검사를 마치는 것보다 지속 양성 상태에서 벗어나는 것이었습니다.
따라서 향후 중재는 해당 에피소드를 P에서 N1으로 이동시키는 능력을 먼저 평가할 가치가 있습니다. 첫 적격 음성까지의 시간은 유용한 중간 평가변수가 될 수 있지만, 이를 국소 제거 확인으로 표현해서는 안 됩니다.
음성 1회는 취약했고, 음성 2회는 훨씬 강한 근거였습니다
N1에서는 55.64%가 N2로 진행하고 44.00%가 P로 재설정되었습니다. 다중비교를 고려하면 여전히 팽팽한 상태였습니다.
N2에서는 73.10%가 C에 도달하고 26.90%가 P로 재설정되었으며, 이 차이는 강하게 지지되었습니다. 따라서 세 번째 음성은 중복된 행정검사가 아니라 확인 가치가 남아 있는 검사였습니다.
제거 시작일과 제거 확인일은 서로 다릅니다
원 논문의 제거까지 시간은 나중에 성공한 3회 연속 음성의 첫 음성일로 소급됩니다. 이는 회고적 시작일입니다. N1 시점에는 앞으로 두 번째와 세 번째 음성이 이어질지 알 수 없습니다.
4상태 재구성은 미래 정보가 과거의 실시간 예측으로 누출되는 문제를 방지하며, 확인 연속검사가 완성되기 전에 첫 음성을 “제거”로 표현하는 오류를 줄입니다.
제거에는 여러 번의 시도가 필요한 경우가 많았습니다
중단 없는 P → N1 → N2 → C 경로의 단순 곱은 6.82%였지만, 적격 시퀀스 중 37.61%는 결국 C에 도달했습니다. 양성 지속, 실패한 음성 연속, 재설정, 이후 재시도가 제거 과정의 본질적 일부였습니다.
P
양성
16.78%
→
N1
첫 음성
55.64%
→
N2
연속 2회 음성
73.10%
→
C
국소 제거 확인
P 유지 83.22% · N1에서 P로 재설정 44.00% · N2에서 P로 재설정 26.90% · 관찰된 C → P 전이 42.03%
III. 조작적 제거 뒤에도 검출 재양성이 흔했습니다
C는 영구적인 생물학적 박멸을 의미하지 않았습니다. 재양성 추정치는 분석단위와 제거 후 감시 지속 여부에 따라 크게 달라졌습니다.
평가변수
추정치
정확한 임상 표현
C → P 전이
29/69 = 42.03%
C에서 관찰된 다음 전이 중 양성이 나온 비율
제거 후 추적된 시퀀스
21/39 = 53.85%
제거 후 추적된 시퀀스 중 한 번 이상 재양성이 검출된 비율
모든 제거 시퀀스
21/88 = 23.86%
제거 후 추적이 없었던 시퀀스까지 분모에 포함한 비율
제거 후 추적된 환자
17/27 = 62.96%
제거 후 추적된 환자 중 하나 이상의 재양성 시퀀스가 있었던 비율
모든 제거 환자
17/43 = 39.53%
하나 이상의 국소 C가 있었던 전체 환자 중 검출 재양성이 있었던 비율
첫 재양성까지의 관찰 중앙값은 11일, 사분위범위는 7–31일이었습니다. 제거 직후가 중요한 시기일 수 있지만, 현재 데이터는 검출한계 아래의 지속 집락화, 간헐적 배출, 검체 변동, 검사 변동, 실제 재획득을 구분하지 못합니다. 따라서 제거 후 검출 재양성이라는 표현이 가장 방어적입니다.
제거 직후 표적 재검사는 전향적으로 평가할 가치가 있는 가설입니다. 현재 결과만으로 새로운 의무 감시 일정을 확정할 수는 없습니다.
IV. 현재 상태는 사망보다 먼저 제거될 가능성에 대한 동적 정보를 제공합니다
연속시간 경쟁위험 모형에서 사망보다 먼저 국소 C가 확인될 확률은 음성 근거가 누적될수록 증가했습니다.
시작 상태
사망 전 C
C 전 사망
임상적 해석
P
42.07%
57.93%
국소 제거 확인까지 상당한 거리가 남은 상태입니다
N1
57.81%
42.19%
균형은 개선되지만 P로의 재설정이 여전히 흔합니다
N2
77.73%
22.27%
적합된 모형에서는 국소 C 확인이 훨씬 더 유리합니다
P에서 시작한 90일 추정치는 C 확인 10.59%, 사망 23.41%, P/N1/N2 미해결 상태 66.00%였습니다. 단기적으로 가장 흔한 결과는 제거도 사망도 아니라 장기간 미해결 상태였습니다.
이는 에피소드 단위 모형 추정치이며 임상적 격리해제 임계값이 아닙니다. 또한 N2가 구조적으로 C에 더 가깝기 때문에 확률이 높아질 수 있으므로, N1이나 N2가 사망을 예방한다고 해석해서는 안 됩니다. 상태별 사망위험에 대한 별도 근거도 혼재했습니다.
V. 사망 전 음성 배양의 급증은 입증되지 않았습니다
사전에 정한 마지막 4주 가설은 음성이었습니다
완전한 짝지은 환자 16명에서 마지막 4주와 직전 4주의 차이는 −4.78퍼센트포인트, 95% 구간은 −12.60에서 +3.81, 단측 P 값은 0.8671이었습니다.
사망 정렬 말기 구간은 이전 구간보다 낮았습니다
짝지은 말기-이전 차이는 −8.53퍼센트포인트, 95% 구간은 −14.08에서 −2.69였습니다.
사망 정렬 궤적과 독립 중도절단 궤적은 달랐습니다
조정 베이지안 모형에서 사망 정렬 음성확률은 22.62%에서 20.38%로 변했지만, 독립 중도절단 정렬 음성확률은 24.06%에서 33.25%로 변했습니다. 사망군 변화가 중도절단군 변화보다 클 확률은 1.10%에 불과했습니다.
채취되지 않았거나 채취할 수 없었던 검체를 음성으로 바꾸어서는 안 됩니다. 현재 분석은 사망 직전 자연스러운 탈집락화를 지지하지 않으며, 지속 양성이 사망을 일으킨다는 인과관계도 입증하지 않습니다.
VI. 숨은 전이 이질성은 존재하지만, 아직 검증된 생물학적 아형은 아닙니다
양성·음성 이분형 모형은 불충분했습니다
병원체–부위별 전이 균질성과 1차 무기억성 가정이 강하게 위반되었습니다. 이분형 HMM이 이분형 Markov 모형보다 상대적으로 적합도가 좋았지만, 어느 모형도 모든 관찰 패턴을 재현하지 못했습니다. 현재 결과만이 아니라 이전 연속 이력이 중요하다는 뜻입니다.
2체제 switching HMM이 적합도와 환자 분리 예측을 개선했습니다
전이
낮은 진행 체제
높은 진행 체제
해석
P → N1
7.55%
54.34%
같은 P라도 음성 연속과정을 시작할 가능성이 크게 다를 수 있습니다
N1 → N2
28.51%
69.77%
첫 음성이 어떤 상황에서는 불안정하지만 다른 상황에서는 강한 전진 신호일 수 있습니다
N2 → C
42.07%
79.30%
연속 2회 음성 뒤에도 주변 전이 맥락이 중요합니다
C → C
16.46%
61.45%
국소 C 이후의 지속성도 체제에 따라 크게 다릅니다
이 체제는 측정되지 않은 항생제 노출, 기구, 감염원 조절, 기도 기구, 상처 부담, 미생물군 교란, 균량, 기타 시간가변 요인을 반영할 수 있습니다. 다만 수학적 전이 양상일 뿐 “두 종류의 환자”는 아닙니다.
생물학적 HMM은 근거 단계의 순서를 지지하지만 실제 개인 확률을 검증하지 않습니다
평균 잠재 집락화 확률은 P 92.53%, N1 25.50%, N2 2.96%, C 0.94%로 감소했습니다. 이는 음성 근거가 누적된다는 해석과 일관됩니다.
그러나 초기 상태 가정에 매우 민감했고, 민감도와 특이도는 외부 미생물학적 금표준 없이 추정된 방출확률입니다. 이를 검증된 검사실 특성이나 개별 환자의 실제 생물학적 확률로 표현해서는 안 됩니다.
VII. 현재 임상적으로 말할 수 있는 내용과 피해야 할 주장
현재 방어 가능한 내용
가설 생성 단계
현재 데이터가 지지하지 않는 주장
첫 적격 음성이 가장 큰 병목입니다
중재가 P를 N1으로 이동시키는지 평가할 수 있습니다
첫 음성이 제거와 같습니다
N1은 취약하고 N2는 훨씬 유리하며 세 번째 음성이 확인 역할을 합니다
검증 후 현재 상태를 동적 상담이나 위험도별 추적에 활용할 수 있습니다
N2가 사망률을 인과적으로 낮춥니다
제거 후 추적된 단위에서 재양성이 흔하고 이른 경우가 많습니다
제거 직후 표적 재검사를 전향적으로 평가할 가치가 있습니다
새로운 보편적 재검사 일정이 이미 정당화됩니다
사망 전 마지막 4주 음성 증가는 입증되지 않았습니다
검체 확보 가능성과 배양 결과를 함께 모형화할 수 있습니다
지속 양성이 임박한 사망을 예측하거나 일으킵니다
두 잠재 전이 체제가 보완 모형을 개선했습니다
임상 공변량이 체제 소속이나 전환을 설명할 수 있습니다
두 체제가 검증된 생물학적 표현형입니다
국소 C는 환자–병원체–부위의 조작적 종결점입니다
30·60·90일 지속 국소 제거를 연구해야 합니다
국소 C가 영구 박멸, PLC, PtLC, 자동 격리해제와 같습니다
VIII. 데이터 경계와 가장 강한 논문 프레이밍
문제
중요한 이유
2,772 → 2,722 → 2,703 → 2,673 관찰
원고 총계, 병원체 소계, 파싱 관찰, 조정 관찰은 서로 다른 단계입니다. 최초 50건의 원고 내부 산술 차이와 8건의 오류 수준 준비 이슈는 원자료 감사를 받아야 합니다.
서로 다른 분석단위
관찰, 전이, 국소 시퀀스, 환자, 사망 연계 에피소드는 바꿔 쓸 수 없습니다. 분자·분모·고유 환자 수·군집화 방법을 계속 표시해야 합니다.
선택적인 제거 후 추적
재양성 비율은 검출 의존적입니다. 지속 제거를 위해 반복사건 또는 다상태 시간-사건 모형이 필요합니다.
정보성 관찰 과정
검체 채취 강도가 관찰 음성과 연관되었습니다. 검사를 많이 하면 제거가 생긴다고 해석해서는 안 됩니다.
동시 집락화 정보의 미활용
원 논문의 CRE 중심 비대칭 네트워크는 병원체–부위 에피소드를 독립적으로만 보는 대신 결합 다병원체 상태모형으로 확장할 근거가 됩니다.
논문에 사용할 수 있는 방어적 종합 문장: 이 전향적 장기요양병원 코호트에서 MDRO 제거는 이력 의존적인 다단계 과정으로 나타났습니다. 가장 큰 장벽은 적격 음성 연속과정을 시작하는 것이었습니다. 음성 근거가 누적될수록 국소 제거 확인이 점차 유리해졌지만, 중간 상태는 P로 재설정될 수 있었고 조작적 제거 뒤에도 검출 재양성이 나타날 수 있었습니다. 사망은 제거와 경쟁했고, P에서 시작한 에피소드의 대부분은 90일에도 미해결 상태였으며, 사망 직전 배양 음성 증가는 입증되지 않았습니다. 이러한 결과는 연속 근거, 재설정, 제거 후 추적, 경쟁 사건을 명시적으로 해석해야 함을 지지하지만, 국소 제거를 영구 박멸·환자 수준 제거·사망률의 인과적 감소와 동일시해서는 안 됩니다.
따라서 두 번째 연구의 가장 강한 정체성은 동적 감시 및 제거 경로 분석이며, 격리해제 기준의 검증으로 표현하는 것은 아직 이릅니다. 가장 가치 있는 숨은 정보는 배양 결과의 연속 이력 자체가 임상적 의미를 가진다는 점입니다.
Written on August 5, 2026
What the nGeneMDRO analysis actually shows: a clinically readable interpretation (Written August 6, 2026)
This report is not a single statistical result. It is a layered investigation of the same longitudinal culture dataset from several different perspectives: operational clearance, biological-state inference, post-clearance re-positivity, terminal culture patterns, competing risks, and latent transition heterogeneity.
The most defensible overall conclusion is that MDRO clearance in this cohort was a difficult, multistep, history-dependent process. The main obstacle was beginning a qualifying negative run. Once negative evidence accumulated, confirmed local clearance became progressively more likely, but positive resets remained possible, operational clearance was not always durable, and many episodes remained unresolved before death or censoring.
I. The central findings at a glance
Question
Main result
Evidence judgment
Clinical meaning
Where is the principal clearance bottleneck?
P → N1 occurred in only 16.78% of next included cultures
Strongly supported descriptively
Beginning a negative run was much harder than completing it after evidence had accumulated
Does the third negative still matter?
N2 → C occurred in 73.10%, while N2 → P still occurred in 26.90%
Strong support after multiplicity adjustment
Two negatives were highly favorable but did not make the third confirmation redundant
Is local clearance durable?
C → P occurred in 29/69 transitions; detected recurrence involved 21 sequences and 17 patients
Descriptive with major follow-up caution
Operational clearance did not guarantee permanent non-detection
Do negative cultures surge before death?
Final four weeks versus previous four weeks: −4.78 percentage points, one-sided P = 0.8671
Not supported
The data do not support spontaneous terminal decolonization
Does current clearance stage inform the path before death?
Estimated C-before-Death probability increased from 42.07% in P to 77.73% in N2
Model-based, supported with caution
Accumulated negative evidence identifies a more favorable process position, not a causal protective treatment
Is one homogeneous transition process sufficient?
A two-hidden-regime four-state HMM improved penalized fit and held-out prediction
Supported as a complementary model
The same observed stage can occur under markedly different latent transition contexts
Most robust
Four-state operational transitions, the N2 confirmation advantage, outcome linkage, and the existence of latent transition heterogeneity
Useful with caution
Competing-risk probabilities, terminal mixed models, recurrence estimates, and pathogen-site subgroup patterns
Exploratory
Absolute biological HMM probabilities, state-specific Death effects, Bayesian predictive superiority, and exact hidden-regime interpretation
Not supported
A final-four-week negative surge, a distinct terminal change point, or adequacy of a pooled simple two-state model
II. The analysis begins with an unresolved data-reconciliation problem
Four observation totals appear in the report. They describe different provenance stages and must not be treated as interchangeable denominators.
2,772
Manuscript-stated total
−50
→
2,722
Manuscript pathogen subtotal
−19
→
2,703
Explicit parsed observations
−30
→
2,673
Prepared observations
321
Prepared local sequences
→
234
Four-state eligible sequences
→
198
Hidden-regime comparison sequences
Difference
Meaning
Required action
2,772 − 2,722 = 50
The manuscript’s pathogen-specific counts do not sum to its stated total
Identify whether the 50 represent duplicated episodes, non-target cultures, unsorted categories, or arithmetic error
2,722 − 2,703 = 19
The manuscript subtotal and parser reconstruction differ
Map each difference to an exact source file and source line
2,703 − 2,673 = 30
Same-day reconciliation or sequence preparation removed or merged observations
Retain a row-level audit showing the deterministic rule applied to every observation
8 sequence-preparation errors
The pipeline completed, but error-level preparation notices remain
Resolve these before describing the dataset as publication-locked
These discrepancies do not automatically invalidate every derived result. They do mean that the numerical analysis should remain provisional until the 50, 19, and 30 observation differences and the eight preparation errors are fully source-traceable.
III. The four-state model gives the clearest clinical story
The primary analytical unit is one patient × pathogen × anatomic site. The model does not begin with “cleared” versus “not cleared.” It follows how evidence accumulates.
P
Positive
16.78%
→
N1
First negative
55.64%
→
N2
Two negatives
73.10%
→
C
Local confirmation
Positive reset remains possible from N1, N2, and C.
The first negative is the dominant bottleneck
Of 1,782 counted transitions from P, 83.22% remained P and only 16.78% reached N1. The principal difficulty was therefore not obtaining the third negative. It was escaping persistent positivity sufficiently to begin a qualifying negative run.
Clinically, this suggests that the most sensitive intermediate research endpoint may be time to the first qualifying negative. That endpoint must still be kept separate from confirmed clearance.
One negative is fragile
After N1, progression to N2 occurred in 55.64%, while a positive reset occurred in 44.00%. The numerical advantage favored progression, but the familywise-adjusted comparison remained inconclusive.
A single negative should therefore be interpreted as an encouraging but unstable result, not as evidence that colonization has ended.
Two negatives are strong evidence, but not completion
After N2, 73.10% confirmed C and 26.90% reset to P. This difference remained strongly supported after multiplicity adjustment.
The third negative is therefore not merely administrative repetition. Approximately one quarter of N2-origin transitions still failed through positive reset.
Clearance often requires more than one attempt
The product of one uninterrupted P → N1 → N2 → C path was only 6.82%, whereas 37.61% of eligible sequences eventually reached C.
The difference represents persistence, failed negative runs, resets, and later successful attempts. The immediate clean-run probability and eventual clearance fraction are therefore fundamentally different endpoints.
Clearance onset and confirmation occur at different times
The median time to any N1 was 20 days. The median onset date of a subsequently successful clearance run was 34.5 days, and median confirmation occurred at 48.5 days.
An early N1 can fail and reset. The clearance-onset date is identified retrospectively as the first negative of the run that ultimately succeeds.
IV. Serial negatives carry biological information, but the exact HMM probabilities are not yet clinically transportable
The biological HMM asks whether an unobserved state of “colonized” or “clear” might underlie the observed positive and negative cultures. Its most important result is the ordering of the stages, not the precise individual probability assigned to each stage.
Model specification
P
N1
N2
C
Interpretation
Positive-conditioned biological HMM
92.53%
25.50%
2.96%
0.94%
Primary design-aligned profile used in the executive summary
Stratified pooled linkage HMM
89.14%
13.23%
1.22%
0.29%
A different fitted specification with fallback and stratification rules
What is robust
Both specifications show a steep monotone decline from P through C. Patient-cluster bootstrap support for the P > N1 > N2 > C gradient was 100%. Serial negative evidence therefore contains substantially more information than a single binary result.
What is fragile
The positive-conditioned HMM was highly sensitive to the initial hidden-state assumption. A stationary initial-state profile had a BIC approximately 27 points lower than the design-aligned positive-conditioned profile. Multiple likelihood modes and boundary-like emission estimates were also present.
What should not be said
N1 should not be described as proving that an individual has exactly a 25.50% chance of remaining colonized. Likewise, the fitted sensitivity and specificity are not validated laboratory test characteristics. They may absorb biological heterogeneity, specimen quality, intermittent shedding, and observation processes.
The defensible biological statement is that the sequence P → N1 → N2 → C represents progressively stronger evidence against continuing colonization. The exact percentages remain model-dependent and exploratory.
V. The pathogen-site map suggests that initiation and confirmation are different ecological problems
The following domain coverage map shows two conditional probabilities within each pathogen-site stratum:
P → N1, followed by N2 → C. These are not cumulative probabilities and do not use the same denominator.
Site
CRE
VRE
MRPA
MRAB
Stool
9.5% → 59.4%
67 sequences
15.6% → 65.0%
36 sequences
Protocol omitted
Protocol omitted
Urine
18.8% → 74.1%
35 sequences
41.1% → 95.0%
23 sequences
32.5% → 80.0%
10 sequences
100.0% → NE
1 sequence · sparse
Sputum
15.7% → 62.5%
31 sequences
NE
66.7% → 60.0%
9 sequences
67.9% → 90.9%
14 sequences
Wound
16.7% → 100.0%
2 sequences · sparse
NE
27.3% → 100.0%
3 sequences · sparse
34.8% → 100.0%
3 sequences · sparse
Blood
NE
NE
NE
NE
The most stable ecological pattern is that CRE stool had exceptional difficulty beginning a negative run. VRE urine and sputum isolates of MRPA or MRAB appeared more likely to begin negative runs, but several non-CRE cells were small.
The map also illustrates why initiation and completion should be modeled separately. A stratum may have difficulty reaching N1 but still have a favorable N2 → C probability once two negatives have accumulated.
A displayed 100% in a sparse cell is not evidence of certainty. It means that every observed event in a very small denominator followed that path. Sparse cells should remain descriptive or be partially pooled in a hierarchical model.
VI. Post-clearance re-positivity is clinically important, but its percentage depends on who was actually followed
Endpoint
Result
What the denominator means
Transition-level C → P
29/69 = 42.03%
Positive result among counted next transitions observed from C
Followed cleared sequences
21/39 = 53.85%
At least one detected recurrence among sequences with post-C transition follow-up
All cleared sequences
21/88 = 23.86%
Includes 49 sequences with no counted post-C transition
Followed cleared patients
17/27 = 62.96%
At least one recurrent sequence among patients who continued post-C surveillance
All cleared patients
17/43 = 39.53%
Includes 16 patients without a counted post-C transition
Why there were 106 clearance episodes but only 88 cleared sequences
A local sequence can reach C, return to P, and later reach C again. Therefore, 88 sequences generated 106 clearance episodes.
Why followed-only and all-cleared percentages differ
A recurrence cannot be detected without another culture. The followed-only percentage can be high because monitored sequences have an opportunity to reveal recurrence. The all-cleared percentage can be low because units without follow-up remain in the denominator even though their recurrence status is unknown.
Why the median of 11 days matters
The first detected recurrence occurred at a median of 11 days, with an interquartile range of 7–31 days. This suggests that the early post-C period deserves prospective study.
It does not yet establish a mandatory Day 7, Day 14, or Day 30 surveillance rule because post-C culture timing was not standardized.
What “recurrence” biologically means remains unresolved
A later positive culture could reflect persistent colonization below detection, intermittent shedding, specimen variability, laboratory variation, or true reacquisition. The safest term is detected post-clearance re-positivity.
VII. The terminal analyses do not support a negative-culture surge before death
Analysis
Death-aligned result
Interpretation
Complete final 4 versus previous 4 weeks
−4.78 pp; 95% interval −12.60 to +3.81; one-sided P = 0.8671
No supported increase; the point estimate was lower
Complete final 8 versus previous 8 weeks
+3.85 pp; 95% interval −11.98 to +20.94; one-sided P = 0.3355
A higher point estimate without confirmatory support
Broader paired terminal 1–4 versus earlier 5–12 weeks
−8.53 pp; 95% interval −14.08 to −2.69
Observed negativity was lower in the terminal window
Adjusted Bayesian death window
22.62% earlier versus 20.38% terminal; change −2.24 pp
P(terminal increase) = 22.85%
Adjusted Bayesian independent-censoring window
24.06% earlier versus 33.25% terminal; change +9.19 pp
P(terminal increase) = 99.33%
Death change minus censoring change
−11.44 pp; 95% credible interval −20.99 to −1.76
P(death change exceeds censoring change) = 1.10%
The confirmatory question was answered negatively
The prespecified final-four-week test did not show an increase in negative cultures. The evidence points away from a claim that residents spontaneously decolonize immediately before death.
The death-versus-censoring contrast is more informative than the death curve alone
The adjusted death-aligned curve remained flat to mildly decreasing, while the independent-censoring curve increased toward its endpoint. The difference-in-differences therefore suggests that the death-aligned trajectory was relatively less favorable.
Different paired sample sizes are not a contradiction
The strict final-four-week confirmatory test required complete D−1–4 and D−5–8 windows and retained only 16 paired patients. The broader terminal-window comparison used patients represented somewhere in weeks 1–4 and weeks 5–12 and retained 28. They answer related but non-identical questions.
Aggregate and paired estimates can point in different directions
In the censoring cohort, unpaired aggregate patient-weighted negativity was higher in the terminal window, but the paired mean difference was −2.05 percentage points. This occurs because the terminal-only and earlier-only patients differ from the subset represented in both periods.
The terminal models describe culture negativity, not mortality causation
Pathogen and site odds ratios in these models refer to the odds of an observed negative culture. They do not estimate pathogen-specific mortality hazards. Likewise, the association between sampling intensity and negativity should not be interpreted as evidence that additional testing causes clearance.
The GAMM had better patient-separated predictive log loss than the linear mixed model, 0.623 versus 0.646, but substantially worse BIC because of its greater flexibility. It is therefore preferable for describing nonlinear curve shape, not because every spline coefficient has a direct clinical interpretation.
VIII. Competing-risk analysis shows that the dominant short-term outcome is often “still unresolved”
The competing-risk model follows one local episode until confirmed C, Death, or right censoring occurs first. It included 199 episodes from 77 patients: 54 C-first, 83 Death-first, and 62 right-censored.
Starting state
Eventual C before Death
Eventual Death before C
90-day C
90-day Death
Still P/N1/N2 at 90 days
P
42.07%
57.93%
10.59%
23.41%
66.00%
N1
57.81%
42.19%
34.13%
18.11%
47.76%
N2
77.73%
22.27%
65.25%
10.04%
24.71%
Accumulated negative evidence moves the episode closer to C
The fitted probability of C occurring before Death increased from P through N2. This is consistent with the operational construction: N2 is already one qualifying negative away from C.
Most P episodes remain unresolved at 90 days
Starting from P, only 10.59% were estimated to reach C by 90 days, while 23.41% reached Death and 66.00% remained in P, N1, or N2. Reporting only clearance or only death would conceal the largest category.
The state gradient is not a causal treatment effect
Reaching N1 or N2 was not randomly assigned. Episodes that survived long enough and produced qualifying negatives were selected into those states. The model does not show that inducing N2 would itself reduce mortality.
The rates are continuous-time approximations
Transition intensities account for unequal exposure time, but exact state-change times remain interval-censored between culture dates. The generator assumes approximately homogeneous rates within each state.
IX. Two latent transition regimes reveal hidden process heterogeneity
A two-regime switching HMM was the preferred complementary model. It retained the observed P, N1, N2, and C states but allowed their transition probabilities to depend on an unobserved transition context.
Transition
Lower-progression regime
Higher-progression regime
Clinical reading
P → N1
7.55%
54.34%
The same observed P state can conceal a seven-fold difference in starting a negative run
N1 → N2
28.51%
69.77%
A first negative may be highly fragile or strongly progressive depending on latent context
N2 → C
42.07%
79.30%
Even after two negatives, transition context remains important
C → C
16.46%
61.45%
Durability after operational clearance differs markedly
Observed occupancy
68.36%
31.64%
The lower-progression context occupied more of the observed sequence time
Both regimes were highly persistent: the lower-progression regime remained itself with probability 97.24% per transition, and the higher-progression regime with probability 99.67%. The process therefore did not appear to switch randomly at every culture.
Possible underlying explanations include antibiotic exposure, device status, source control, respiratory instrumentation, wound burden, organism load, specimen quality, microbiome disruption, or other unmeasured time-varying factors.
These are latent transition regimes, not proven biological stages and not two permanent patient types. A patient or sequence may change regime over time.
The two-regime model was preferred by AICc and BIC and improved patient-level cross-validated log loss by roughly 4% over the operational MM. A four-regime model achieved a slightly lower cross-validation loss, but the difference from the two-regime model was below 1%, while complexity and BIC deteriorated substantially. The five-regime model did not converge.
X. The models answer different questions and should not compete for one universal winner
Model
Best use
Current judgment
Main limitation
Four-State Operational MM
Direct interpretation of the three-negative protocol and positive resets
Primary model
Uses next-observation transitions and does not explain hidden heterogeneity
Two-regime Four-State HMM
Transition prediction and latent process heterogeneity
Supported complementary model
Regimes lack measured clinical labels and need repeated validation
Biological two-state HMM
Relating observed evidence stages to an inferred colonized/clear process
Exploratory
Initial-state sensitivity, local modes, boundary emissions, and no external gold standard
Continuous-time competing-risk multi-state model
C-before-Death, Death-before-C, and time-horizon probabilities
Supported with caution
Homogeneous-rate approximation and interval-censored transition times
GAMM terminal model
Nonlinear death- and censoring-aligned culture trajectories
Preferred for predictive curve shape
Wide uncertainty and no standalone interpretation of spline coefficients
Five-State MM with Death
Testing whether Death transitions differ by P, N1, N2, and C
Mixed evidence
AICc and nominal likelihood-ratio evidence favor complexity, while BIC and Bayes factors favor the shared-Death model
Bayesian versus plug-in prediction
Testing predictive shrinkage within the Five-State structure
Inconclusive
Log-loss interval crossed zero and Brier improvement was negligible
Simple pooled two-state MM/HMM
Reference and assumption-failure demonstration
Neither adequate
Pathogen-site heterogeneity, memorylessness violation, and failed predictive checks
The Five-State Death comparison is a useful example of why one criterion is insufficient. The Five-State model had lower AICc and a nominal likelihood-ratio P value of 0.0105, but its BIC was worse, its exact Bayes factor strongly favored the simpler shared-Death model, and the conclusion changed with the prior scale. State-specific Death probabilities therefore remain unconfirmed.
XI. What this derived study adds to the original manuscript
Original study contribution
Derived nGeneMDRO contribution
New clinical insight
SAC, PAC, PLC, and PtLC clearance hierarchy
P, N1, N2, and C longitudinal evidence states within each local sequence
Clearance can be decomposed into initiation, continuation, confirmation, reset, and recurrence
Time to retrospectively confirmed clearance
Real-time stage at each culture
The first negative should not be back-labeled as clearance before future confirmation exists
Site and pathogen effects on clearance
Stage-specific transition patterns and latent transition regimes
The factors governing escape from P may differ from those governing completion after N1 or N2
Mortality analyzed by site, pathogen, age, and sex
C-versus-Death competing first-event pathways and reverse-time terminal analyses
Many episodes remain unresolved, and no terminal negative surge was demonstrated
Clearance as an endpoint
Detected post-C re-positivity with explicit transition, sequence, and patient denominators
Operational clearance and durable non-detection are different outcomes
Conditional co-colonization matrix
No completed Clinical Bayesian Network result appears in this export
Temporal direction, base-rate enrichment, and conditional dependencies still require a separate appended analysis
XII. The Clinical Bayesian Network results are missing from this exported report
The section manifest contains 36 analytical sections, but none is a completed Clinical Bayesian Networks section. The report mentions Bayesian networks only in the future-method roadmap.
Therefore, the following cannot be judged from this document:
Which Bayesian Network parents were selected for P → N1, N1 → N2, N2 → C, C → P, mortality, or recurrence
Reservoir-discordance and stool-gate audit results from the new module
This likely means either that the canonical report was generated before the Clinical Bayesian Network module was run, or that the canonical report renderer has not yet appended its Markdown output. The network analysis should be re-run and incorporated before the exported document is described as containing the complete Bayesian Network findings.
The original manuscript’s asymmetric co-colonization finding remains part of the source study. However, this export does not newly validate temporal direction, independent enrichment over the high CRE base rate, or an incident number-needed-to-test strategy.
XIII. Clinically defensible statements and overstatements to avoid
Clinically defensible
Not established by the current analysis
The first qualifying negative is the main operational bottleneck
A first negative means that colonization has cleared
Negative evidence becomes progressively more convincing from N1 to N2 to C
N1, N2, or C is identical to a directly observed biological state
The third negative retains meaningful confirmation value
Two negatives are sufficient for universal isolation release
Local C may be followed by detected re-positivity
C means permanent eradication, PLC, or PtLC
Post-C detection often occurred early among followed units
The exact recurrence incidence is 62.96% for all patients
No final-four-week increase in observed negativity was supported
Persistent positivity causes death or predicts imminent death
Current operational state is associated with different modeled C-before-Death pathways
Moving a patient experimentally to N2 would causally reduce mortality
Two latent transition regimes improve the complementary model
The cohort consists of two validated biological patient phenotypes
The biological HMM supports the ordering of serial evidence
Its sensitivity and specificity are validated culture-test characteristics
XIV. The highest-priority next analyses
Lock the data provenance before expanding the models
Every one of the 50 manuscript arithmetic differences, 19 parser differences, 30 reconciliation reductions, and eight preparation errors should be represented in a deterministic audit table.
Repeat patient-level cross-validation for HMM order stability
The two-regime choice is sensible, but the present order comparison used one patient-fold assignment. Repeated folds should quantify how often two, three, or four regimes win.
Jointly model recurrence and the observation process
Post-C culture timing and recurrence detection should be modeled together, or surveillance should be prospectively standardized at prespecified days.
Add time-varying clinical variables
Antibiotic exposure, devices, tracheostomy, feeding route, pressure wounds, source control, acute infection, organism burden, and functional status could explain the hidden transition regimes.
Model PLC and PtLC as slowest-reservoir processes
Aggregate clearance should be analyzed as the maximum of several local clearance times, with site-specific frailty and interval censoring rather than as an ordinary average.
Convert co-colonization into a temporal risk-set analysis
For each first MRPA, MRAB, or VRE detection, the analysis should distinguish CRE already known, CRE detected on the same date, and incident CRE detected afterward. Conditional yield, lift over baseline prevalence, and number needed to test should remain separate.
Perform temporal or external validation
The most persuasive validation would apply the fixed definitions and fitted models to a later SSCH cohort or an independent LTCF without re-estimating every rule from the same data.
XV. Final clinical synthesis
In this prospective LTCF dataset, MDRO clearance behaved as a multistep evidence process rather than a single conversion event. Persistent positivity was difficult to escape, the first negative remained fragile, two negatives provided strong but incomplete evidence, and the third negative retained meaningful confirmation value. Local operational clearance was sometimes followed by detected re-positivity, particularly among units that continued surveillance. No increase in negative cultures was demonstrated during the final four weeks before death. Competing-risk models showed that accumulated negative evidence identified a more favorable pathway toward confirmed local clearance, but many episodes remained unresolved and no causal mortality effect was established. A two-hidden-regime HMM supported substantial latent transition heterogeneity, although those regimes cannot yet be assigned biological identities. The four-state operational model should remain primary, while the hidden-regime, biological HMM, terminal, Bayesian, and competing-risk models should remain complementary analyses with their distinct limitations visible.
nGeneMDRO 분석이 실제로 보여주는 것: 임상적으로 이해하기 쉬운 해석
이 보고서는 하나의 통계 결과가 아닙니다. 동일한 장기 배양자료를 조작적 제거, 생물학적 잠재상태, 제거 후 재양성, 사망 전 배양 변화, 경쟁위험, 숨은 전이 이질성이라는 서로 다른 관점에서 분석한 다층 연구입니다.
가장 방어적인 종합 결론은 다음과 같습니다. 이 코호트에서 MDRO 제거는 어렵고, 여러 단계를 거치며, 과거 배양 이력에 의존하는 과정이었습니다. 가장 큰 장벽은 적격 음성 연속과정을 시작하는 것이었습니다. 음성 근거가 누적될수록 국소 제거 확인 가능성이 높아졌지만, 양성 재설정은 계속 가능했고, 조작적 제거가 항상 지속되지는 않았으며, 사망이나 중도절단 전까지 해결되지 않은 에피소드가 많았습니다.
I. 핵심 결과 한눈에 보기
질문
주요 결과
근거 판단
임상적 의미
제거의 가장 큰 병목은 어디인가?
다음 포함 배양에서 P → N1은 16.78%에 불과
기술적으로 강하게 지지
음성 근거가 쌓인 뒤 제거를 완성하는 것보다 음성 연속과정을 시작하는 것이 훨씬 어려움
세 번째 음성도 의미가 있는가?
N2 → C는 73.10%, N2 → P는 여전히 26.90%
다중비교 보정 후 강한 지지
음성 2회는 매우 유리하지만 세 번째 확인검사가 불필요해지는 것은 아님
국소 제거는 지속되는가?
C → P는 29/69 전이, 검출 재양성은 21개 시퀀스와 17명 환자에서 확인
추적관찰 편향이 큰 기술적 결과
조작적 제거가 영구적인 비검출을 보장하지 않음
사망 직전 음성 배양이 급증하는가?
마지막 4주 대 직전 4주: −4.78퍼센트포인트, 단측 P = 0.8671
지지되지 않음
사망 직전 자연스럽게 탈집락화된다는 가설을 지지하지 않음
현재 제거 단계가 사망 전 경로에 정보를 주는가?
C-before-Death 추정확률이 P의 42.07%에서 N2의 77.73%로 증가
모형 기반, 주의하여 지지
음성 근거 누적은 더 유리한 과정 위치를 뜻하지만 인과적 보호치료를 뜻하지 않음
하나의 균질한 전이과정으로 충분한가?
2개 잠재 체제 4상태 HMM이 벌점 적합도와 외부 환자 예측을 개선
보완모형으로 지지
동일한 관찰 단계에도 크게 다른 숨은 전이 맥락이 존재할 수 있음
가장 견고한 결과
4상태 조작적 전이, N2 이후 확인 우위, 결과자료 연결, 잠재 전이 이질성의 존재
주의하여 활용할 결과
경쟁위험 확률, 말기 혼합모형, 재양성 비율, 병원체–부위 하위군 결과
탐색적 결과
생물학적 HMM 절대확률, 상태별 사망효과, Bayesian 예측 우월성, 잠재 체제의 정확한 생물학적 의미
지지되지 않은 결과
마지막 4주의 음성 급증, 뚜렷한 말기 변화점, 단순 통합 2상태 모형의 적합성
II. 분석은 아직 해결되지 않은 데이터 조정 문제에서 시작합니다
보고서에는 네 가지 관찰 수가 나타납니다. 각각은 서로 다른 자료 provenance 단계를 뜻하므로 동일한 분모처럼 사용해서는 안 됩니다.
2,772
원고 기재 총계
−50
→
2,722
원고 병원체 소계
−19
→
2,703
명시적으로 파싱된 관찰
−30
→
2,673
준비 완료 관찰
321
준비된 국소 시퀀스
→
234
4상태 적격 시퀀스
→
198
잠재 체제 비교 시퀀스
차이
의미
필요한 조치
2,772 − 2,722 = 50
원고의 병원체별 수를 합해도 원고 총계와 맞지 않음
50건이 중복, 비대상 배양, 미분류 항목 또는 산술오류인지 확인
2,722 − 2,703 = 19
원고 소계와 parser 재구성 결과가 다름
각 차이를 정확한 원자료 파일과 줄에 연결
2,703 − 2,673 = 30
동일일자 조정이나 시퀀스 준비 과정에서 관찰이 제거 또는 병합됨
모든 관찰에 적용된 결정론적 규칙을 행 단위 감사표로 보존
시퀀스 준비 오류 8건
파이프라인은 완료되었지만 오류 수준 알림이 남아 있음
출판용 고정 데이터셋이라고 표현하기 전에 해결
이러한 차이가 모든 파생 결과를 자동으로 무효화하는 것은 아닙니다. 다만 50건, 19건, 30건의 차이와 8건의 준비 오류가 모두 원자료 수준에서 추적될 때까지 수치적 해석은 잠정적이어야 합니다.
III. 4상태 모형이 가장 명확한 임상적 이야기를 제공합니다
주 분석단위는 한 환자 × 병원체 × 해부학적 부위입니다. 이 모형은 처음부터 “제거”와 “비제거”로 나누지 않고, 근거가 어떻게 누적되는지를 추적합니다.
P
양성
16.78%
→
N1
첫 음성
55.64%
→
N2
연속 2회 음성
73.10%
→
C
국소 확인
N1, N2, C 이후에도 양성 재설정이 가능합니다.
첫 음성이 가장 큰 병목입니다
P에서 계산된 1,782회 전이 중 83.22%는 P에 남았고, N1에 도달한 비율은 16.78%뿐이었습니다. 가장 큰 어려움은 세 번째 음성을 얻는 것이 아니라, 지속 양성에서 벗어나 적격 음성 연속과정을 시작하는 것이었습니다.
따라서 향후 연구에서 민감한 중간 평가변수는 첫 적격 음성까지의 시간이 될 수 있습니다. 그러나 이 평가변수를 제거 확인과 동일시해서는 안 됩니다.
음성 1회는 취약합니다
N1 이후 N2 진행은 55.64%, 양성 재설정은 44.00%였습니다. 수치적으로는 진행이 우세했지만, 가족단위 다중비교 보정 후에는 확정적 차이로 남지 않았습니다.
따라서 한 번의 음성은 긍정적이지만 불안정한 결과이며, 집락화가 끝났다는 근거는 아닙니다.
음성 2회는 강한 근거이지만 완성은 아닙니다
N2 이후 73.10%가 C를 확인했고, 26.90%는 P로 재설정되었습니다. 이 차이는 다중비교 보정 후에도 강하게 지지되었습니다.
세 번째 음성은 단순한 행정적 반복이 아닙니다. N2에서 시작한 전이의 약 4분의 1은 양성 재설정으로 실패했습니다.
제거에는 여러 차례의 시도가 필요한 경우가 많습니다
중단 없는 P → N1 → N2 → C 경로의 단순 곱은 6.82%였지만, 적격 시퀀스 중 최종적으로 C에 도달한 비율은 37.61%였습니다.
이 차이는 양성 지속, 실패한 음성 연속, 재설정, 이후 재시도를 반영합니다. 즉시 성공경로 확률과 최종 제거 비율은 전혀 다른 평가변수입니다.
제거 시작일과 제거 확인일은 다릅니다
어떤 N1이든 처음 도달하기까지의 중앙값은 20일이었습니다. 나중에 성공한 제거 연속과정의 시작일 중앙값은 34.5일, 확인일 중앙값은 48.5일이었습니다.
초기 N1은 실패하고 재설정될 수 있습니다. 제거 시작일은 나중에 성공한 연속 음성의 첫 음성일로 회고적으로 정해집니다.
IV. 연속 음성은 생물학적 정보를 담지만 HMM의 정확한 확률을 그대로 임상에 적용하기에는 이릅니다
생물학적 HMM은 관찰된 양성·음성 배양 아래에 보이지 않는 “집락화” 또는 “제거” 상태가 존재한다고 가정합니다. 가장 중요한 결과는 각 단계의 순서이며, 개별 단계에 부여된 정확한 확률이 아닙니다.
모형 설정
P
N1
N2
C
해석
양성 조건부 생물학적 HMM
92.53%
25.50%
2.96%
0.94%
Executive summary에 사용된 설계 정렬 주 분석 profile
층화 pooled linkage HMM
89.14%
13.23%
1.22%
0.29%
층화와 fallback 규칙이 다른 별도 적합 모형
견고한 부분
두 설정 모두 P에서 C로 갈수록 잠재 집락화 확률이 급격히 감소합니다. P > N1 > N2 > C 순서에 대한 환자 군집 bootstrap 지지는 100%였습니다. 연속 음성 이력은 단일 양성·음성 결과보다 훨씬 많은 정보를 담습니다.
취약한 부분
양성 조건부 HMM은 초기 잠재상태 가정에 매우 민감했습니다. 정상상태 초기 profile은 설계 정렬 양성 조건부 profile보다 BIC가 약 27 낮았습니다. 여러 likelihood mode와 경계에 가까운 방출확률도 확인되었습니다.
피해야 할 표현
N1 환자에게 실제 집락화 확률이 정확히 25.50%라고 표현해서는 안 됩니다. 적합된 민감도와 특이도 역시 검증된 검사실 성능이 아닙니다. 생물학적 이질성, 검체 질, 간헐적 배출, 관찰과정을 함께 흡수할 수 있습니다.
방어 가능한 생물학적 문장은 P → N1 → N2 → C가 지속 집락화에 반대되는 근거가 점차 강해지는 순서라는 것입니다. 정확한 수치는 모형 의존적이며 탐색적입니다.
V. 병원체–부위 지도를 보면 음성 시작과 제거 확인이 서로 다른 생태학적 문제임을 알 수 있습니다
아래 domain coverage map의 각 칸에는 두 조건부 확률이 표시됩니다.
앞은 P → N1, 뒤는 N2 → C입니다. 두 수치는 누적확률이 아니며 분모도 다릅니다.
부위
CRE
VRE
MRPA
MRAB
대변
9.5% → 59.4%
67개 시퀀스
15.6% → 65.0%
36개 시퀀스
Protocol상 생략
Protocol상 생략
소변
18.8% → 74.1%
35개 시퀀스
41.1% → 95.0%
23개 시퀀스
32.5% → 80.0%
10개 시퀀스
100.0% → NE
1개 시퀀스 · 희소
객담
15.7% → 62.5%
31개 시퀀스
NE
66.7% → 60.0%
9개 시퀀스
67.9% → 90.9%
14개 시퀀스
상처
16.7% → 100.0%
2개 시퀀스 · 희소
NE
27.3% → 100.0%
3개 시퀀스 · 희소
34.8% → 100.0%
3개 시퀀스 · 희소
혈액
NE
NE
NE
NE
가장 안정적인 생태학적 패턴은 CRE 대변이 음성 연속과정을 시작하기 특히 어려웠다는 점입니다. VRE 소변과 MRPA·MRAB 객담은 음성 연속과정을 더 자주 시작한 것으로 보이지만, 일부 비CRE 칸은 표본이 작습니다.
이 지도는 시작과 완성을 따로 분석해야 하는 이유도 보여줍니다. 어떤 층은 N1 도달이 어렵지만, 두 번의 음성이 이미 쌓인 뒤에는 N2 → C가 유리할 수 있습니다.
희소 칸의 100%는 확실성을 뜻하지 않습니다. 매우 작은 분모에서 관찰된 사건이 모두 같은 경로를 따랐다는 뜻입니다. 희소 칸은 기술적으로만 제시하거나 계층모형으로 부분 pooling해야 합니다.
VI. 제거 후 재양성은 임상적으로 중요하지만 실제 추적을 받은 대상에 따라 비율이 달라집니다
평가변수
결과
분모의 의미
전이 수준 C → P
29/69 = 42.03%
C에서 관찰된 계산 가능한 다음 전이 중 양성 결과
추적된 제거 시퀀스
21/39 = 53.85%
C 이후 전이 추적이 있었던 시퀀스 중 한 번 이상 재양성
모든 제거 시퀀스
21/88 = 23.86%
C 이후 계산 가능한 전이가 없었던 49개 시퀀스까지 포함
추적된 제거 환자
17/27 = 62.96%
C 이후 감시가 지속된 환자 중 하나 이상의 재양성 시퀀스
모든 제거 환자
17/43 = 39.53%
C 이후 계산 가능한 전이가 없었던 16명까지 포함
제거 시퀀스는 88개인데 제거 episode가 106개인 이유
하나의 국소 시퀀스가 C에 도달한 뒤 P로 돌아가고, 나중에 다시 C에 도달할 수 있습니다. 따라서 88개 시퀀스에서 106회의 제거 episode가 생성되었습니다.
추적군 비율과 전체 제거군 비율이 다른 이유
재양성은 추가 배양이 있어야 검출됩니다. 추적군 비율은 재양성을 발견할 기회가 있기 때문에 높을 수 있습니다. 전체 제거군 비율은 추적이 없는 대상까지 분모에 남기 때문에 낮아질 수 있지만, 이들의 실제 재양성 상태는 알 수 없습니다.
중앙값 11일이 중요한 이유
첫 검출 재양성까지의 중앙값은 11일, 사분위범위는 7–31일이었습니다. 제거 직후의 초기 기간을 전향적으로 연구할 필요가 있음을 시사합니다.
다만 C 이후 검사시점이 표준화되지 않았으므로 Day 7, Day 14, Day 30 의무검사 일정을 확정하는 근거는 아직 아닙니다.
생물학적으로 재양성이 무엇을 뜻하는지는 아직 해결되지 않았습니다
후속 양성은 검출한계 아래의 지속 집락화, 간헐적 배출, 검체 변동, 검사실 변동, 실제 재획득 중 어느 것이든 가능할 수 있습니다. 가장 안전한 용어는 제거 후 검출 재양성입니다.
VII. 말기 분석은 사망 전 음성 배양 급증을 지지하지 않습니다
분석
사망 정렬 결과
해석
완전한 마지막 4주 대 직전 4주
−4.78 pp; 95% 구간 −12.60에서 +3.81; 단측 P = 0.8671
증가가 지지되지 않으며 점추정치는 오히려 낮음
완전한 마지막 8주 대 직전 8주
+3.85 pp; 95% 구간 −11.98에서 +20.94; 단측 P = 0.3355
점추정치는 높지만 확인적 근거는 없음
넓은 범위의 말기 1–4주 대 이전 5–12주 짝비교
−8.53 pp; 95% 구간 −14.08에서 −2.69
말기 구간에서 관찰 음성률이 낮음
조정 Bayesian 사망 구간
이전 22.62% 대 말기 20.38%; 변화 −2.24 pp
P(말기 증가) = 22.85%
조정 Bayesian 독립 중도절단 구간
이전 24.06% 대 말기 33.25%; 변화 +9.19 pp
P(말기 증가) = 99.33%
사망 변화 − 중도절단 변화
−11.44 pp; 95% 신용구간 −20.99에서 −1.76
P(사망군 변화가 더 큼) = 1.10%
사전에 정한 확인적 질문의 답은 음성이었습니다
마지막 4주 확인검정에서는 음성 배양 증가가 나타나지 않았습니다. 사망 직전 자연스럽게 탈집락화된다는 주장은 지지되지 않습니다.
사망곡선 단독보다 사망 대 중도절단 비교가 더 중요합니다
조정된 사망 정렬곡선은 평탄하거나 완만하게 감소했지만, 독립 중도절단곡선은 endpoint에 가까워질수록 증가했습니다. Difference-in-differences는 사망 정렬 궤적이 상대적으로 불리했음을 시사합니다.
짝지은 환자 수가 다른 것은 모순이 아닙니다
엄격한 마지막 4주 확인검정은 D−1–4와 D−5–8의 모든 주가 완전해야 하므로 16명만 포함했습니다. 더 넓은 말기 구간 비교는 1–4주와 5–12주에 일부라도 관찰된 환자를 사용하여 28명이 포함되었습니다. 서로 관련되지만 동일한 질문은 아닙니다.
전체 평균과 짝비교가 다른 방향을 보일 수 있습니다
중도절단군에서는 짝을 맞추지 않은 전체 환자 가중 음성률이 말기 구간에서 높았지만, 짝지은 평균차이는 −2.05퍼센트포인트였습니다. 말기만 관찰된 환자와 이전 구간만 관찰된 환자의 구성이 두 구간 모두 관찰된 환자와 다르기 때문입니다.
말기 모형은 배양 음성률을 설명하며 사망 원인을 설명하지 않습니다
이 모형의 병원체와 부위 odds ratio는 관찰된 음성 배양의 odds입니다. 병원체별 사망위험을 뜻하지 않습니다. 검사 강도와 음성률의 연관성 역시 검사를 많이 하면 제거가 일어난다는 인과관계가 아닙니다.
GAMM은 선형 혼합모형보다 환자 분리 예측 log loss가 0.623 대 0.646으로 좋았지만, 더 복잡하여 BIC는 훨씬 나빴습니다. 따라서 비선형 곡선 모양을 설명하는 데 유리한 것이며, 각 spline 계수에 직접적인 임상 의미가 있다는 뜻은 아닙니다.
VIII. 경쟁위험 분석에서는 단기적으로 가장 흔한 결과가 ‘아직 미해결’입니다
경쟁위험 모형은 하나의 국소 episode를 C 확인, 사망, 또는 우측 중도절단 중 먼저 발생한 사건까지 추적합니다. 77명에서 199개 episode가 포함되었으며, C-first 54개, Death-first 83개, 우측 중도절단 62개였습니다.
시작 상태
최종적으로 사망 전 C
최종적으로 C 전 사망
90일 C
90일 사망
90일에도 P/N1/N2
P
42.07%
57.93%
10.59%
23.41%
66.00%
N1
57.81%
42.19%
34.13%
18.11%
47.76%
N2
77.73%
22.27%
65.25%
10.04%
24.71%
음성 근거가 누적될수록 C에 가까워집니다
사망보다 먼저 C가 발생할 적합 확률은 P에서 N2로 갈수록 높아졌습니다. N2는 이미 C까지 적격 음성 1회만 남은 상태이므로 조작적 정의와 일관됩니다.
P에서 시작한 episode 대부분은 90일에도 미해결입니다
P에서 시작하면 90일 C는 10.59%, 사망은 23.41%, P·N1·N2에 남아 있는 비율은 66.00%였습니다. 제거 또는 사망 하나만 보고하면 가장 큰 범주가 사라집니다.
상태 차이는 인과적 치료효과가 아닙니다
N1이나 N2 도달은 무작위 배정이 아닙니다. 충분히 생존하고 적격 음성을 만들어낸 episode가 해당 상태로 선택됩니다. N2를 유도하면 그 자체로 사망률이 감소한다는 결과가 아닙니다.
전이율은 연속시간 근사치입니다
전이강도는 서로 다른 노출시간을 반영하지만 실제 상태변화 시점은 배양일 사이에 구간 중도절단되어 있습니다. Generator는 각 상태 안에서 대략 균질한 전이율을 가정합니다.
IX. 두 잠재 전이 체제는 숨겨진 과정 이질성을 보여줍니다
2체제 switching HMM이 가장 적절한 보완모형으로 선택되었습니다. 관찰되는 P, N1, N2, C는 그대로 유지하면서, 이들 사이 전이확률이 보이지 않는 전이 맥락에 따라 달라지도록 했습니다.
전이
낮은 진행 체제
높은 진행 체제
임상적 해석
P → N1
7.55%
54.34%
같은 P 상태 안에도 음성 연속과정 시작 가능성이 약 7배 다를 수 있음
N1 → N2
28.51%
69.77%
첫 음성이 매우 취약한 맥락과 강하게 진행하는 맥락이 존재
N2 → C
42.07%
79.30%
음성 2회 이후에도 전이 맥락이 중요
C → C
16.46%
61.45%
조작적 제거 이후 지속성도 크게 다름
관찰 점유율
68.36%
31.64%
관찰된 시퀀스 시간 중 낮은 진행 맥락이 더 많았음
두 체제 모두 매우 지속적이었습니다. 낮은 진행 체제는 한 전이 뒤에도 같은 체제에 남을 확률이 97.24%, 높은 진행 체제는 99.67%였습니다. 매 배양 때마다 무작위로 바뀌는 과정은 아니었습니다.
항생제 노출, 기구, 감염원 조절, 기관절개나 호흡기 처치, 상처 부담, 균량, 검체 질, 미생물군 교란, 기타 측정되지 않은 시간가변 요인이 잠재 체제를 설명할 수 있습니다.
이는 잠재 전이 체제이지 검증된 생물학적 단계가 아니며, 환자를 영구적으로 두 유형으로 나누는 분류도 아닙니다. 한 환자나 시퀀스가 시간에 따라 체제를 바꿀 수 있습니다.
2체제 모형은 AICc와 BIC에서 우세했고, 조작적 MM보다 환자 수준 교차검증 log loss를 약 4% 개선했습니다. 4체제 모형이 약간 더 낮은 교차검증 손실을 보였지만 2체제와의 차이는 1% 미만이었고 복잡도와 BIC가 크게 악화되었습니다. 5체제 모형은 수렴하지 않았습니다.
5상태 사망 비교는 하나의 기준만으로 결론을 내리면 안 되는 대표적인 예입니다. 5상태 모형은 AICc가 낮고 명목 likelihood-ratio P 값이 0.0105였지만, BIC는 더 나빴고 정확 Bayes factor는 단순 공통 사망모형을 강하게 선호했으며 prior scale에 따라 결론이 달라졌습니다. 상태별 사망확률 차이는 아직 확인되지 않았습니다.
XI. 이 파생 연구가 원 논문에 추가하는 내용
원 연구의 기여
nGeneMDRO 파생분석의 기여
새로운 임상적 통찰
SAC, PAC, PLC, PtLC 제거 계층
각 국소 시퀀스 안의 P, N1, N2, C 종적 근거상태
제거를 시작, 지속, 확인, 재설정, 재양성으로 분해할 수 있음
회고적으로 확인된 제거까지 시간
각 배양시점의 실시간 단계
미래 확인이 존재하기 전에 첫 음성을 제거로 소급해서는 안 됨
부위와 병원체가 제거에 미치는 영향
단계별 전이 양상과 잠재 전이 체제
P에서 벗어나는 기전과 N1·N2 이후 완성 기전이 다를 수 있음
부위, 병원체, 연령, 성별에 따른 사망 분석
C 대 사망 경쟁 첫 사건과 역시간 말기 분석
미해결 episode가 많고 사망 직전 음성 급증은 확인되지 않음
제거를 하나의 endpoint로 평가
전이·시퀀스·환자 분모를 구분한 C 이후 재양성
조작적 제거와 지속 비검출은 서로 다른 결과
조건부 동시집락화 행렬
현재 export에는 완료된 Clinical Bayesian Network 결과가 없음
시간적 방향성, 기저율 보정 농축, 조건부 의존성은 별도 분석을 추가해야 함
XII. 이 export에는 Clinical Bayesian Network 결과가 포함되어 있지 않습니다
Section manifest에는 36개 분석 section이 있지만, 완료된 Clinical Bayesian Networks section은 없습니다. Bayesian Network는 future-method roadmap에만 언급되어 있습니다.
따라서 이 문서만으로는 다음을 판단할 수 없습니다.
P → N1, N1 → N2, N2 → C, C → P, 사망, 재양성 network에서 선택된 부모변수
환자 군집 bootstrap edge stability
BDeu prior sensitivity
환자 분리 Bayesian Network 교차검증
CRE 높은 기저율을 보정한 동시집락화 농축도
새 모듈의 reservoir discordance와 stool-gate 감사결과
Canonical report가 Clinical Bayesian Network 모듈 실행 전에 생성되었거나, canonical renderer가 아직 해당 Markdown 출력을 붙이지 않은 것으로 볼 수 있습니다. Network 분석을 다시 실행하여 포함하기 전에는 이 export를 전체 Bayesian Network 결과까지 포함한 완전한 보고서라고 표현하기 어렵습니다.
원 논문의 비대칭 동시집락화 결과는 원 연구의 일부로 유지됩니다. 그러나 이 export는 시간적 선후관계, 높은 CRE 기저율을 넘는 독립적 농축, incident number-needed-to-test 전략을 새롭게 검증하지는 않았습니다.
XIII. 임상적으로 방어 가능한 문장과 피해야 할 과장
임상적으로 방어 가능한 내용
현재 분석으로 입증되지 않은 내용
첫 적격 음성이 가장 큰 조작적 병목입니다
첫 음성이 곧 집락화 제거를 의미합니다
N1, N2, C로 갈수록 음성 근거가 점차 강해집니다
N1, N2, C가 직접 관찰한 생물학적 상태와 같습니다
세 번째 음성은 의미 있는 확인가치를 유지합니다
음성 2회만으로 모든 상황에서 격리해제가 가능합니다
국소 C 뒤에도 검출 재양성이 나타날 수 있습니다
C가 영구 박멸, PLC 또는 PtLC와 같습니다
추적된 대상에서는 C 이후 재양성이 이른 경우가 많았습니다
모든 환자의 정확한 재양성 발생률이 62.96%입니다
사망 전 마지막 4주의 관찰 음성률 증가는 지지되지 않았습니다
지속 양성이 사망을 일으키거나 임박한 사망을 예측합니다
현재 조작상태에 따라 C-before-Death 모형경로가 달랐습니다
환자를 N2로 이동시키면 인과적으로 사망률이 낮아집니다
두 잠재 전이 체제가 보완모형을 개선했습니다
코호트가 두 개의 검증된 생물학적 환자형으로 구성됩니다
생물학적 HMM은 연속 근거의 순서를 지지합니다
HMM의 민감도와 특이도가 검증된 배양검사 성능입니다
XIV. 우선순위가 가장 높은 다음 분석
모형을 더 확장하기 전에 데이터 provenance를 고정해야 합니다
원고 산술 차이 50건, parser 차이 19건, 조정 감소 30건, 준비 오류 8건을 모두 결정론적 감사표에 표시해야 합니다.
HMM 체제 수 안정성을 반복 환자 교차검증으로 확인해야 합니다
2체제 선택은 합리적이지만 현재 비교는 하나의 환자 fold 배정에 의존합니다. 반복 fold에서 2·3·4체제가 각각 얼마나 자주 승리하는지 확인해야 합니다.
재양성과 관찰과정을 함께 모형화해야 합니다
C 이후 배양시점과 재양성 검출을 joint model로 분석하거나 전향적으로 정해진 날짜에 감시해야 합니다.
시간가변 임상변수를 추가해야 합니다
항생제 노출, 기구, 기관절개, 영양경로, 욕창, 감염원 조절, 활동성 감염, 균량, 기능상태가 잠재 전이 체제를 설명할 수 있습니다.
PLC와 PtLC를 가장 느린 저장소 과정으로 모형화해야 합니다
집계 제거는 여러 국소 제거시간의 평균이 아니라 최댓값에 가까운 과정입니다. 부위별 frailty와 구간 중도절단을 포함한 weakest-link 모형이 적절합니다.
동시집락화를 시간적 risk-set 분석으로 전환해야 합니다
MRPA, MRAB, VRE 첫 검출마다 이미 알려진 CRE, 같은 날 검출된 CRE, 이후 새로 검출된 CRE를 분리해야 합니다. 조건부 yield, 기저 유병률 대비 lift, number needed to test는 각각 따로 제시해야 합니다.
시간적 또는 외부검증이 필요합니다
가장 설득력 있는 검증은 같은 규칙과 모형을 이후 SSCH 코호트 또는 독립 LTCF에 적용하고, 동일 자료에서 모든 규칙을 다시 추정하지 않는 방식입니다.
XV. 최종 임상적 종합
이 전향적 LTCF 자료에서 MDRO 제거는 단일 전환사건이 아니라 다단계 근거과정으로 나타났습니다. 지속 양성에서 벗어나기 어려웠고, 첫 음성은 취약했으며, 음성 2회는 강하지만 불완전한 근거였고, 세 번째 음성은 의미 있는 확인가치를 유지했습니다. 국소 조작적 제거 뒤에도 재양성이 검출될 수 있었으며, 특히 감시가 계속된 대상에서 자주 확인되었습니다. 사망 전 마지막 4주에 음성 배양이 증가한다는 근거는 없었습니다. 경쟁위험 모형에서 음성 근거가 누적될수록 국소 제거 확인으로 향하는 경로가 유리해졌지만, 미해결 episode가 많았고 인과적 사망효과는 입증되지 않았습니다. 2체제 HMM은 상당한 잠재 전이 이질성을 지지했지만 각 체제에 생물학적 정체성을 부여할 수는 없습니다. 4상태 조작적 모형을 주 분석으로 유지하고, 잠재 체제 HMM, 생물학적 HMM, 말기 모형, Bayesian 분석, 경쟁위험 모형은 각각의 한계를 명시한 보완분석으로 사용해야 합니다.
Written on August 6, 2026
Decoding nGeneMDRO: the hidden clinical structure of longitudinal MDRO clearance (Written August 6, 2026)
The central clinical finding is not simply that some patients cleared MDROs and others did not. The dataset reveals a multistep process: persistent positivity is difficult to escape, the first negative is fragile, two negatives are strongly favorable but incomplete, the third negative remains meaningful, local clearance can be followed by detected re-positivity, and many episodes remain unresolved before death or censoring.
98
Residents
Prospective LTCF cohort
2,703
Parsed cultures
Explicit site-specific results
234
Four-state sequences
Patient × pathogen × site
16.78%
P → N1
Main operational bottleneck
73.10%
N2 → C
Third negative confirms C
11 days
First re-positivity
Median among detected events
Most robust
Four-state transitions, N2 confirmation value, complete outcome linkage, and latent transition heterogeneity
Supported with caution
Competing-risk probabilities, terminal mixed models, recurrence estimates, and subgroup patterns
Exploratory
Absolute biological HMM probabilities, state-specific death effects, and exact hidden-regime interpretation
Not supported
A final-four-week negative surge, a distinct terminal change point, or adequacy of the simple pooled two-state family
I. The entire study can be read as one connected clinical map
Raw cultures
Positive and negative results
→
Operational history
P → N1 → N2 → C
→
Hidden biology
Colonized versus clear
→
Competing outcomes
C, Death, or unresolved
→
Hidden regimes
Low versus high progression
Vertical hierarchy
Local site
↓
All sites for one pathogen
↓
All pathogens for one patient
Horizontal trajectory
P
↓ first qualifying negative
N1
↓ second qualifying negative
N2
↓ third qualifying negative
C
Competing clinical pathway
Current state
↙ ↓ ↘
Confirmed C Death Still unresolved
Analytical layer
Main question
Best-supported interpretation
What it cannot establish
Four-State Operational MM
How does evidence progress or reset?
Primary protocol-aligned model
True biological eradication
Biological HMM
What hidden colonization state may underlie the cultures?
Serial negatives provide progressively stronger evidence
Validated individual colonization probability
Recurrence analysis
How often is positivity detected after C?
Re-positivity is common among followed units
True recurrence incidence without standardized follow-up
Terminal analysis
Does negativity rise near death?
No supported final-four-week surge
That persistent positivity causes death
Competing-risk model
Does C or Death occur first?
Current stage changes the fitted pathway
A causal benefit of reaching N1 or N2
Switching HMM
Are there hidden transition contexts?
Two regimes improve fit and prediction
Two permanent biological patient types
II. The data pipeline is informative, but it is not yet fully publication-locked
2,772
Manuscript total
−50
internal arithmetic gap
2,722
Pathogen subtotal
−19
reconstruction gap
2,703
Parsed observations
−30
reconciliation reduction
2,673
Prepared observations
321 prepared patient–pathogen–site sequences
234 observed-positive four-state sequences
87 excluded from the observed-positive analysis
198 hidden-regime comparison sequences
36 additional short sequences excluded
Source inconsistency
The manuscript total and its pathogen subtotal differ by 50.
Parser discrepancy
Nineteen observations separate the manuscript subtotal from parser reconstruction.
Preparation reduction
Thirty parsed observations were merged or removed by reconciliation rules.
Error-level notices
Eight sequence-preparation errors remain visible in the report.
Audit question
Current answer
Publication requirement
Can every manuscript count be reproduced?
No; a 50-observation internal gap remains
Identify the exact category or arithmetic source
Can every parser difference be traced?
Not yet demonstrated in this report
Map all 19 differences to exact files and source lines
Can every removed or merged observation be explained?
Deterministic reconciliation is reported, but the row-level audit is not shown here
Retain the rule and provenance for all 30 observations
Did the analyses execute?
Yes
Execution completeness must remain separate from data-audit completeness
The numerical patterns are coherent enough for secondary analysis, but final publication claims should remain provisional until the 50, 19, and 30 observation differences and the eight preparation errors are individually reconciled.
III. The four-state pathway reveals where clearance succeeds and fails
P
Positive status
16.78%
qualifying negative →
N1
First negative
55.64%
second negative →
N2
Two negatives
73.10%
third negative →
C
Local confirmation
P remains P 83.22%
N1 resets to P 44.00%
N2 resets to P 26.90%
C returns to P 42.03%
Primary bottleneck
Leaving P is difficult. Only 299 of 1,782 P-origin transitions began a negative run.
Fragile evidence
After N1, progression and positive reset remain nearly balanced.
Strong evidence
N2 strongly favors C, but more than one quarter still reset.
Operational confirmation
C is meaningful, but it is not automatically permanent eradication.
Median time landmarks
Baseline positive Day 0
→
First N1 of any attempt 20 days
→
N2 38 days
→
Successful-run onset 34.5 days
→
C confirmation 48.5 days
The first negative is the main operational bottleneck
Persistent positivity dominates the early process. A future intervention may therefore show its earliest useful signal by shortening time to the first qualifying negative, even when confirmed clearance remains uncommon.
The third negative still contributes real information
N2 was highly favorable, but 26.90% of N2-origin transitions returned to P. The third negative is therefore not redundant within this dataset.
Immediate success and eventual success are different endpoints
The uninterrupted P → N1 → N2 → C product was only 6.82%, whereas 37.61% of sequences eventually reached C after allowing repeated attempts and resets.
IV. The pathogen–site landscape shows that starting clearance and completing clearance are different problems
Each cell below contains two conditional probabilities:
P → N1 followed by N2 → C. The first measures difficulty beginning a negative run. The second measures confirmation after two qualifying negatives have already accumulated.
Site
CRE
VRE
MRPA
MRAB
Stool
9.5% → 59.4% 67 sequences
15.6% → 65.0% 36 sequences
Protocol omitted
Protocol omitted
Urine
18.8% → 74.1% 35 sequences
41.1% → 95.0% 23 sequences
32.5% → 80.0% 10 sequences
100.0% → NE 1 sequence · sparse
Sputum
15.7% → 62.5% 31 sequences
NE
66.7% → 60.0% 9 sequences
67.9% → 90.9% 14 sequences
Wound
16.7% → 100.0% 2 sequences · sparse
NE
27.3% → 100.0% 3 sequences · sparse
34.8% → 100.0% 3 sequences · sparse
Blood
NE
NE
NE
NE
Left side of the map
Difficult to leave P
Right side of the map
Easier to begin a negative run
Lower part of the map
N2 confirmation remains uncertain
Upper part of the map
N2 usually progresses to C
CRE stool
The clearest initiation bottleneck: P → N1 only 9.5%.
VRE urine
Relatively favorable initiation and very high N2 confirmation.
MRPA/MRAB sputum
High P → N1 estimates, but subgroup sizes remain modest.
Sparse 100% cells
A value of 100% with one to three sequences is not evidence of certainty.
V. The original study and the derived analysis describe complementary dimensions
Original study: reservoir axis
Stool: 181 days
Sputum: 78 days
Urine: 47 days
Wound: 28 days
The gastrointestinal reservoir was the most persistent.
Original study: pathogen axis
CRE: 159 days
VRE: 61 days
MRPA: 27 days
MRAB: 28 days
CRE was the most persistent target organism.
Derived study: trajectory axis
P → N1: initiation
N1 → N2: continuation
N2 → C: confirmation
C → P: detected re-positivity
The culture history reveals where each local episode succeeds or fails.
Original manuscript
Derived nGeneMDRO analysis
Combined clinical interpretation
Stool and CRE cleared most slowly
CRE stool also had the lowest observed P → N1 probability
The major difficulty may be initiating a negative run within a persistent intestinal CRE reservoir
Clearance was independent of age and sex
Operational stages primarily reflected culture history and reservoir context
Local ecological and microbiological factors may dominate general demographic factors for clearance
Mortality depended on site and host factors rather than pathogen identity
No terminal negative surge was demonstrated
Colonization dynamics and mortality mechanisms should not be collapsed into one pathogen-centered explanation
PLC and PtLC aggregate several local units
Local sequences often follow different trajectories
Aggregate clearance behaves as a slowest-reservoir or weakest-link process
Why patient-level clearance can remain delayed
Urine cleared
+
Sputum cleared
+
Stool still positive
=
PLC or PtLC remains unresolved
VI. The biological HMM supports an evidence gradient, not a precise bedside probability
Observed operational track
P→N1→N2→C
Directly reconstructed from serial cultures
Hidden biological track
Colonized↔Clear
Inferred, model-dependent, and not directly observed
P
Mean 92.53%
N1
Mean 25.50%
N2
Mean 2.96%
C
Mean 0.94%
Finding
Visual meaning
Clinical conclusion
Monotone P > N1 > N2 > C gradient
Each additional qualifying negative sharply lowers inferred colonization probability
Serial evidence is clinically informative
100% patient-cluster bootstrap support for the gradient
The ordering remained stable under patient-level resampling
The direction of the gradient is robust within the fitted model
Stationary profile BIC 27.08 points lower than the primary profile
The model changes materially when the baseline hidden-state assumption changes
Absolute probabilities remain exploratory
Boundary and local-mode warnings
Different parameter combinations can explain similar culture sequences
HMM sensitivity and specificity are not validated test characteristics
The defensible statement is that P → N1 → N2 → C represents progressively stronger evidence against ongoing colonization. It is not defensible to tell an individual patient that N1 proves an exact 25.50% probability of remaining colonized.
VII. Post-clearance re-positivity must be read through its denominator
Sequence denominator
88 cleared sequences
39 with post-C follow-up
21 with detected recurrence
Patient denominator
43 cleared patients
27 with post-C follow-up
17 with detected recurrence
Observed time to first detected re-positivity
Q1: 7 days
Median: 11 days
Q3: 31 days
Long tail to 301 days
Endpoint
Estimate
Correct interpretation
C → P transition
29/69 = 42.03%
Positive result among counted next transitions observed from C
Followed cleared sequences
21/39 = 53.85%
Detected recurrence among sequences with post-C transition follow-up
All cleared sequences
21/88 = 23.86%
Includes 49 sequences without a counted post-C transition
Followed cleared patients
17/27 = 62.96%
At least one recurrent sequence among followed patients
All cleared patients
17/43 = 39.53%
Includes 16 patients without any counted post-C transition
Pathogen–site
Followed sequences
Recurrent sequences
Recurrence among followed
Median days
CRE · Sputum
4
3
75.00%
8
CRE · Stool
9
5
55.56%
27
CRE · Urine
8
4
50.00%
7
MRPA · Urine
3
2
66.67%
67
VRE · Stool
5
1
20.00%
7
VRE · Urine
7
4
57.14%
29.5
The safest biological term is detected post-clearance re-positivity. The current data cannot distinguish persistent colonization below detection, intermittent shedding, sampling variation, laboratory variation, or true reacquisition.
VIII. The terminal analysis does not show spontaneous decolonization before death
D−12
D−11
D−10
D−9
D−8
D−7
D−6
D−5
D−4
D−3
D−2
D−1
Confirmatory reference window D−5 through D−8
Confirmatory terminal window D−1 through D−4
Terminal question
Patients
Difference
Uncertainty
Conclusion
Final 4 versus previous 4 weeks
16 paired
−4.78 pp
95% interval −12.60 to +3.81; one-sided P 0.8671
No supported increase
Final 8 versus previous 8 weeks
10 paired
+3.85 pp
95% interval −11.98 to +20.94; one-sided P 0.3355
Higher point estimate without confirmation
Terminal 1–4 versus earlier 5–12 weeks
28 paired
−8.53 pp
95% interval −14.08 to −2.69
Lower observed negativity near death
Death change minus censoring change
Adjusted model
−11.44 pp
95% credible interval −20.99 to −1.76
Death-aligned change was less favorable
Not demonstrated
A sudden increase in negative cultures immediately before death
Observed instead
Flat or lower death-aligned negativity, depending on the analytical window
Important comparator
Independent-censoring negativity increased toward its endpoint in the adjusted Bayesian model
Interpretation boundary
These models describe collected cultures, not a causal mechanism of terminal illness
IX. Competing-risk analysis shows why “still unresolved” must remain visible
P
21,714 exposure-days
N1
3,111 exposure-days
N2
2,095 exposure-days
C
Absorbing first event
Death
Competing first event
P → N1: 0.8612 per 100 days
P → Death: 0.3224 per 100 days
N1 → N2: 2.7965 per 100 days
N1 → Death: 0.2893 per 100 days
N2 → C: 2.5776 per 100 days
N2 → Death: 0.1909 per 100 days
Starting state
Eventual C before Death
Eventual Death before C
90-day C
90-day Death
Still unresolved at 90 days
P
42.07%
57.93%
10.59%
23.41%
66.00%
N1
57.81%
42.19%
34.13%
18.11%
47.76%
N2
77.73%
22.27%
65.25%
10.04%
24.71%
Episode unit
199 local episodes from 77 patients
C-first
54 episodes
Death-first
83 episodes
Right-censored
62 episodes
The state gradient is prognostic within the fitted process, not causal. N1 and N2 were not randomly assigned treatments. Episodes had to survive, remain observed, and produce qualifying negatives to enter those states.
X. Two hidden transition regimes expose a major concealed source of heterogeneity
Lower-progression regime
P → N1 7.55%
N1 → N2 28.51%
N2 → C 42.07%
C → C 16.46%
Observed occupancy: 68.36%
Remain in the same regime: 97.24%
Higher-progression regime
P → N1 54.34%
N1 → N2 69.77%
N2 → C 79.30%
C → C 61.45%
Observed occupancy: 31.64%
Remain in the same regime: 99.67%
Model
BIC
CV log loss
Minimum occupancy
Converged
Judgment
Operational MM / 1 regime
2288.97
0.5068
100.00%
Yes
Primary interpretation model
2 regimes
2250.20
0.4862
31.64%
Yes
Recommended complementary model
3 regimes
2321.94
0.4835
27.59%
Yes
More complex, less separated
4 regimes
2415.22
0.4822
13.15%
Yes
Best single CV split, but less than 1% better than two regimes
5 regimes
2530.28
0.4839
5.15%
No
Should not be selected
The same observed state can have very different futures
P → N1 differed from 7.55% to 54.34% across the two regimes. The label P alone therefore conceals substantial process heterogeneity.
The regimes are persistent rather than rapidly alternating
Both regime self-transition probabilities exceeded 97%, suggesting sustained transition contexts rather than random switching at every culture.
The regimes do not yet have clinical names
Antibiotic exposure, devices, source control, respiratory instrumentation, wound burden, organism load, specimen quality, and microbiome disruption are plausible explanations, but none is established by the current data.
XI. Model selection is a portfolio decision rather than a contest for one universal winner
Clinical question
Preferred model
Why
Evidence boundary
How does the three-negative protocol unfold?
Four-State Operational MM
Directly represents P, N1, N2, C, and positive resets
Primary descriptive and inferential framework
Does hidden transition heterogeneity improve prediction?
Two-regime Four-State HMM
Best balance of AICc, BIC, prediction, occupancy, and separation
Regimes remain unlabeled and observational
What hidden biological state may underlie the cultures?
Biological two-state HMM
Provides a coherent evidence gradient
Highly initial-state sensitive and exploratory
Does C or Death occur first?
Continuous-time competing-risk model
Uses exposure time and competing first events
Homogeneous-rate approximation
Is the terminal trajectory nonlinear?
GAMM
Lower patient-level CV log loss than the linear mixed model
Higher BIC and wide curve uncertainty
Does Death require state-specific probabilities?
No decisive winner
AICc and nominal LR favored five states; BIC and Bayes factors favored shared Death
Mixed evidence
Does Bayesian shrinkage improve prediction?
Not established
Log-loss improvement interval crossed zero; Brier improvement was negligible
Inconclusive
Is a simple pooled positive/negative model sufficient?
No
Homogeneity, memorylessness, and predictive checks failed
Reference model only
Primary
Four-State Operational MM
Complementary
Two-regime switching HMM
Exploratory biology
Two-state biological HMM
Outcome pathway
Competing-risk model
Terminal shape
GAMM and Bayesian model
XII. Clinically defensible claims and claims that remain unsupported
Defensible now
Not established
The first qualifying negative is the main operational bottleneck
A first negative means biological clearance
Serial negatives become progressively more convincing
N1, N2, or C is identical to a directly observed biological state
The third negative retains meaningful confirmation value
Two negatives justify universal isolation release
Local C can be followed by detected re-positivity
Local C equals permanent eradication, PLC, or PtLC
Detected re-positivity often occurred early among followed units
The true recurrence incidence is 62.96% for all cleared patients
No final-four-week negative surge was supported
Persistent positivity causes or reliably predicts imminent death
Current state is associated with different fitted C-versus-Death pathways
Experimentally moving a patient to N2 would causally reduce mortality
Two latent transition regimes improve the complementary model
The cohort consists of two proven biological patient phenotypes
Important reporting gap: Clinical Bayesian Networks are absent from this export
The section manifest contains 36 analytical sections, but no completed Clinical Bayesian Network section. The current document therefore does not show selected Bayesian Network parents, patient-cluster edge stability, BDeu prior sensitivity, patient-separated network validation, base-rate-adjusted co-colonization enrichment, or the new reservoir-discordance audit.
Pathogen
Site
Host factors
Observation process
→
P → N1, N1 → N2, N2 → C, C → P, Death, or fixed-horizon recurrence
XIII. The next research program should target the unresolved mechanisms
Priority 1 · Data lock
Reconcile 50, 19, and 30 observations and review all eight preparation errors.
Priority 2 · Regime stability
Repeat patient-level cross-validation across many fold assignments.
Priority 3 · Observation process
Jointly model post-C culture timing and recurrent positivity.
Which unresolved reservoir controls aggregate clearance?
Targets the site actually delaying isolation release
Temporal co-colonization risk set
Does another MDRO truly enrich future CRE detection?
Provides valid screening yield and number needed to test
Temporal or external validation
Do the findings persist outside the derivation cohort?
Determines transportability and clinical credibility
XIV. Final clinical synthesis
Reservoir and pathogen ecology
→
Difficulty leaving P
→
Fragile N1
→
Strong N2 evidence
→
Local C
Detected re-positivity
↖
Hidden transition regime
↗
Death or unresolved follow-up
In this prospective LTCF dataset, MDRO clearance behaved as a history-dependent multistep process rather than a single conversion event. The principal barrier was initiation of a qualifying negative run. Accumulated negative evidence progressively favored confirmed local clearance, but intermediate states could reset and local C could be followed by detected re-positivity. No increase in negative cultures was demonstrated during the final four weeks before death. Competing-risk models showed that many episodes remained unresolved, while a two-regime HMM revealed substantial hidden transition heterogeneity. The Four-State Operational MM should remain the primary model, with the switching HMM, biological HMM, terminal models, and competing-risk analysis serving distinct complementary roles.
nGeneMDRO 해석: 장기 MDRO 제거과정에 숨어 있던 임상적 구조
핵심 임상결과는 단순히 어떤 환자는 제거되었고 어떤 환자는 제거되지 않았다는 것이 아닙니다. 이 자료에는 다단계 과정이 숨어 있습니다. 지속 양성에서 벗어나기 어렵고, 첫 음성은 취약하며, 음성 2회는 강하지만 불완전한 근거이고, 세 번째 음성은 여전히 의미가 있으며, 국소 제거 뒤에도 재양성이 검출될 수 있고, 사망이나 중도절단 전까지 미해결 상태로 남는 episode가 많았습니다.
98명
연구대상자
전향적 LTCF 코호트
2,703건
파싱 배양관찰
명시적 부위별 결과
234개
4상태 시퀀스
환자 × 병원체 × 부위
16.78%
P → N1
가장 큰 조작적 병목
73.10%
N2 → C
세 번째 음성이 C를 확인
11일
첫 재양성
검출된 사건의 중앙값
가장 견고한 결과
4상태 전이, N2의 확인가치, 완전한 결과연결, 잠재 전이 이질성
주의하여 지지되는 결과
경쟁위험 확률, 말기 혼합모형, 재양성 비율, 하위군 양상
탐색적 결과
생물학적 HMM 절대확률, 상태별 사망효과, 잠재 체제의 정확한 생물학적 의미
지지되지 않은 결과
마지막 4주 음성 급증, 뚜렷한 말기 변화점, 단순 pooled 2상태 모형의 적합성
I. 전체 연구는 하나의 연결된 임상지도로 읽을 수 있습니다
원 배양결과
양성과 음성
→
조작적 이력
P → N1 → N2 → C
→
숨은 생물학
집락화 대 제거
→
경쟁 결과
C, 사망, 미해결
→
숨은 전이 체제
낮은 진행 대 높은 진행
수직 계층
국소 부위
↓
한 병원체의 모든 부위
↓
한 환자의 모든 병원체
수평 궤적
P
↓ 첫 적격 음성
N1
↓ 두 번째 적격 음성
N2
↓ 세 번째 적격 음성
C
경쟁 임상경로
현재 상태
↙ ↓ ↘
C 확인 사망 미해결 지속
분석층
주요 질문
가장 방어적인 해석
입증할 수 없는 내용
4상태 조작적 MM
근거가 어떻게 진행하거나 재설정되는가?
주 protocol 정렬 모형
실제 생물학적 박멸
생물학적 HMM
배양 아래에 어떤 잠재 집락화 상태가 있는가?
연속 음성이 점차 강한 근거를 제공
검증된 개인별 집락화 확률
재양성 분석
C 뒤에 양성이 얼마나 검출되는가?
추적된 대상에서 재양성이 흔함
표준화 추적 없는 실제 발생률
말기 분석
사망 직전 음성률이 증가하는가?
마지막 4주 급증은 지지되지 않음
지속 양성이 사망을 일으킨다는 인과관계
경쟁위험 모형
C와 사망 중 무엇이 먼저 발생하는가?
현재 상태에 따라 적합경로가 달라짐
N1 또는 N2 도달의 인과적 보호효과
Switching HMM
숨은 전이 맥락이 존재하는가?
2개 체제가 적합도와 예측을 개선
두 개의 영구적 생물학적 환자유형
II. 데이터 파이프라인은 유용하지만 아직 출판용으로 완전히 고정되지는 않았습니다
2,772
원고 총계
−50
원고 내부 산술 차이
2,722
병원체 소계
−19
재구성 차이
2,703
파싱 관찰
−30
조정 감소
2,673
준비 완료 관찰
321개 준비 완료 환자–병원체–부위 시퀀스
234개 관찰양성 4상태 시퀀스
87개는 관찰양성 분석에서 제외
198개 잠재 체제 비교 시퀀스
짧은 시퀀스 36개 추가 제외
원자료 불일치
원고 총계와 원고의 병원체 소계가 50건 차이 납니다.
Parser 차이
원고 소계와 parser 재구성 사이에 19건 차이가 있습니다.
준비단계 감소
30건이 동일일자 조정규칙에 따라 제거 또는 병합되었습니다.
오류 수준 알림
시퀀스 준비 오류 8건이 보고서에 남아 있습니다.
감사 질문
현재 답변
출판 전 요구사항
원고의 모든 수를 재현할 수 있는가?
아니며 50건의 내부 차이가 남아 있음
정확한 분류 또는 산술오류 원인을 확인
모든 parser 차이를 추적할 수 있는가?
이 보고서만으로는 아직 확인되지 않음
19건을 정확한 파일과 원자료 줄에 연결
제거 또는 병합된 모든 관찰을 설명할 수 있는가?
결정론적 규칙은 보고되지만 행 단위 감사표는 제시되지 않음
30건 모두의 규칙과 provenance를 보존
분석 실행 자체는 완료되었는가?
예
실행 완료와 데이터 감사 완료를 구분해야 함
파생분석을 수행할 수 있을 정도로 패턴은 일관되지만, 50건·19건·30건의 차이와 준비 오류 8건이 개별적으로 조정되기 전까지 최종 출판수치는 잠정적이어야 합니다.
III. 4상태 경로를 보면 제거가 어디에서 성공하고 실패하는지 보입니다
P
양성 상태
16.78%
적격 음성 →
N1
첫 음성
55.64%
두 번째 음성 →
N2
음성 2회
73.10%
세 번째 음성 →
C
국소 제거 확인
P 유지 83.22%
N1에서 P 재설정 44.00%
N2에서 P 재설정 26.90%
C에서 P 재양성 42.03%
주 병목
P에서 벗어나기 어렵습니다. 1,782회 P 기원 전이 중 299회만 음성 연속과정을 시작했습니다.
취약한 근거
N1 이후 진행과 양성 재설정은 여전히 비슷한 수준입니다.
강한 근거
N2는 C를 강하게 선호하지만 4분의 1 이상이 다시 양성으로 돌아갑니다.
조작적 확인
C는 의미가 있지만 자동으로 영구 박멸을 뜻하지는 않습니다.
중앙값 시간 지표
기저 양성 0일
→
어떤 시도든 첫 N1 20일
→
N2 38일
→
성공 연속과정 시작 34.5일
→
C 확인 48.5일
첫 음성이 가장 큰 조작적 병목입니다
초기 과정은 지속 양성이 지배합니다. 향후 중재는 확인 제거율이 아직 낮더라도 첫 적격 음성까지의 시간을 줄이는지로 초기 효과를 포착할 수 있습니다.
세 번째 음성은 실제 정보를 추가합니다
N2는 매우 유리했지만 N2 기원 전이의 26.90%가 P로 돌아갔습니다. 따라서 이 자료에서 세 번째 음성은 중복검사가 아닙니다.
즉시 성공과 최종 성공은 서로 다른 평가변수입니다
중단 없는 P → N1 → N2 → C 확률은 6.82%였지만, 반복 시도와 재설정을 허용하면 37.61%가 최종적으로 C에 도달했습니다.
IV. 병원체–부위 지도를 보면 음성 시작과 제거 완성이 서로 다른 문제임을 알 수 있습니다
각 칸에는 두 조건부 확률이 표시됩니다.
앞은 P → N1, 뒤는 N2 → C입니다. 첫 수치는 음성 연속과정 시작의 어려움을, 두 번째 수치는 이미 음성 2회가 누적된 뒤 확인 가능성을 보여줍니다.
부위
CRE
VRE
MRPA
MRAB
대변
9.5% → 59.4% 67개 시퀀스
15.6% → 65.0% 36개 시퀀스
Protocol상 생략
Protocol상 생략
소변
18.8% → 74.1% 35개 시퀀스
41.1% → 95.0% 23개 시퀀스
32.5% → 80.0% 10개 시퀀스
100.0% → NE 1개 시퀀스 · 희소
객담
15.7% → 62.5% 31개 시퀀스
NE
66.7% → 60.0% 9개 시퀀스
67.9% → 90.9% 14개 시퀀스
상처
16.7% → 100.0% 2개 시퀀스 · 희소
NE
27.3% → 100.0% 3개 시퀀스 · 희소
34.8% → 100.0% 3개 시퀀스 · 희소
혈액
NE
NE
NE
NE
지도 왼쪽
P에서 벗어나기 어려움
지도 오른쪽
음성 연속과정 시작이 상대적으로 쉬움
지도 아래쪽
N2 이후 확인도 불확실
지도 위쪽
N2가 대체로 C로 진행
CRE 대변
가장 뚜렷한 시작 병목으로 P → N1은 9.5%에 불과합니다.
VRE 소변
시작도 상대적으로 유리하고 N2 확인도 매우 높습니다.
MRPA·MRAB 객담
P → N1 추정치는 높지만 하위군 크기가 크지 않습니다.
희소 100% 칸
1–3개 시퀀스의 100%는 확실성을 뜻하지 않습니다.
V. 원 연구와 파생분석은 서로 보완적인 차원을 설명합니다
원 연구: 저장소 축
대변: 181일
객담: 78일
소변: 47일
상처: 28일
장내 저장소가 가장 오래 지속되었습니다.
원 연구: 병원체 축
CRE: 159일
VRE: 61일
MRPA: 27일
MRAB: 28일
CRE가 가장 오래 지속되는 표적 병원체였습니다.
파생 연구: 궤적 축
P → N1: 시작
N1 → N2: 지속
N2 → C: 확인
C → P: 검출 재양성
각 국소 episode가 어디에서 성공하거나 실패하는지를 보여줍니다.
원 논문
nGeneMDRO 파생분석
통합 임상해석
대변과 CRE의 제거가 가장 느림
CRE 대변에서 P → N1도 가장 낮음
지속적인 장내 CRE 저장소에서 음성 연속과정을 시작하는 것이 특히 어려울 수 있음
제거는 연령·성별과 독립적
조작적 단계는 배양이력과 저장소 맥락을 강하게 반영
제거에는 일반 인구학적 요인보다 국소 생태·미생물학적 요인이 더 중요할 수 있음
사망은 병원체 종류보다 부위와 숙주요인에 연관
사망 직전 음성 급증은 확인되지 않음
집락화 궤적과 사망기전을 하나의 병원체 중심 설명으로 합쳐서는 안 됨
PLC와 PtLC는 여러 국소단위를 집계
국소 시퀀스들은 서로 다른 경로를 따름
집계 제거는 가장 느린 저장소 또는 weakest-link 과정으로 작동
환자수준 제거가 계속 지연될 수 있는 이유
소변 제거
+
객담 제거
+
대변 지속 양성
=
PLC 또는 PtLC 미해결
VI. 생물학적 HMM은 근거의 경사를 지지하지만 정확한 개인 확률을 검증하지는 않습니다
관찰 조작경로
P→N1→N2→C
연속 배양에서 직접 재구성
숨은 생물학 경로
집락화↔제거
모형으로 추정되며 직접 관찰되지 않음
P
평균 92.53%
N1
평균 25.50%
N2
평균 2.96%
C
평균 0.94%
결과
시각적 의미
임상적 결론
P > N1 > N2 > C의 단조경사
적격 음성이 하나씩 쌓일수록 잠재 집락화 확률이 급격히 감소
연속 근거는 임상적으로 유용함
경사에 대한 환자 군집 bootstrap 지지 100%
환자 단위 재표집에도 단계 순서가 유지
적합모형 안에서 방향성은 견고함
정상상태 profile의 BIC가 주 profile보다 27.08 낮음
기저 잠재상태 가정을 바꾸면 모형이 크게 달라짐
절대확률은 탐색적으로 유지해야 함
경계 추정과 여러 likelihood mode
서로 다른 parameter 조합이 비슷한 시퀀스를 설명할 수 있음
HMM 민감도·특이도는 검증된 검사성능이 아님
방어 가능한 문장은 P → N1 → N2 → C가 지속 집락화에 반대되는 근거가 점차 강해지는 순서라는 것입니다. N1 환자의 실제 집락화 확률이 정확히 25.50%라고 설명하는 것은 방어하기 어렵습니다.
VII. 제거 후 재양성은 반드시 분모와 함께 읽어야 합니다
시퀀스 분모
제거 시퀀스 88개
C 이후 추적 39개
재양성 검출 21개
환자 분모
제거 환자 43명
C 이후 추적 27명
재양성 검출 17명
첫 검출 재양성까지의 관찰시간
Q1: 7일
중앙값: 11일
Q3: 31일
최대 301일의 긴 꼬리
평가변수
추정치
정확한 해석
C → P 전이
29/69 = 42.03%
C에서 관찰된 계산 가능한 다음 전이 중 양성
추적된 제거 시퀀스
21/39 = 53.85%
C 이후 전이 추적이 있었던 시퀀스 중 검출 재양성
모든 제거 시퀀스
21/88 = 23.86%
C 이후 계산 가능한 전이가 없었던 49개까지 포함
추적된 제거 환자
17/27 = 62.96%
추적된 환자 중 하나 이상의 재양성 시퀀스
모든 제거 환자
17/43 = 39.53%
C 이후 계산 가능한 전이가 없었던 16명까지 포함
병원체–부위
추적 시퀀스
재양성 시퀀스
추적군 재양성
중앙값 일수
CRE · 객담
4
3
75.00%
8
CRE · 대변
9
5
55.56%
27
CRE · 소변
8
4
50.00%
7
MRPA · 소변
3
2
66.67%
67
VRE · 대변
5
1
20.00%
7
VRE · 소변
7
4
57.14%
29.5
가장 안전한 생물학적 용어는 제거 후 검출 재양성입니다. 현재 자료는 검출한계 아래의 지속 집락화, 간헐적 배출, 검체 변동, 검사실 변동, 실제 재획득을 구분하지 못합니다.
VIII. 말기 분석에서는 사망 전 자연스러운 탈집락화가 나타나지 않았습니다
D−12
D−11
D−10
D−9
D−8
D−7
D−6
D−5
D−4
D−3
D−2
D−1
확인적 기준 구간 D−5부터 D−8
확인적 말기 구간 D−1부터 D−4
말기 질문
환자
차이
불확실성
결론
마지막 4주 대 직전 4주
짝지은 16명
−4.78 pp
95% 구간 −12.60에서 +3.81; 단측 P 0.8671
증가가 지지되지 않음
마지막 8주 대 직전 8주
짝지은 10명
+3.85 pp
95% 구간 −11.98에서 +20.94; 단측 P 0.3355
점추정치만 높고 확인되지 않음
말기 1–4주 대 이전 5–12주
짝지은 28명
−8.53 pp
95% 구간 −14.08에서 −2.69
사망 직전 관찰 음성률이 낮음
사망 변화 − 중도절단 변화
조정모형
−11.44 pp
95% 신용구간 −20.99에서 −1.76
사망 정렬 변화가 상대적으로 불리함
확인되지 않은 내용
사망 직전 음성 배양의 급격한 증가
대신 관찰된 내용
분석구간에 따라 사망 정렬 음성률은 평탄하거나 낮아짐
중요한 비교군
독립 중도절단군은 조정 Bayesian 모형에서 endpoint에 가까워질수록 음성률이 증가
해석 경계
이는 수집된 배양을 설명하는 모형이지 말기질환의 인과기전은 아님
IX. 경쟁위험 분석에서는 ‘아직 미해결’ 범주를 반드시 보여주어야 합니다
P
노출 21,714일
N1
노출 3,111일
N2
노출 2,095일
C
흡수성 첫 사건
사망
경쟁 첫 사건
P → N1: 100일당 0.8612
P → 사망: 100일당 0.3224
N1 → N2: 100일당 2.7965
N1 → 사망: 100일당 0.2893
N2 → C: 100일당 2.5776
N2 → 사망: 100일당 0.1909
시작 상태
최종적으로 사망 전 C
최종적으로 C 전 사망
90일 C
90일 사망
90일에도 미해결
P
42.07%
57.93%
10.59%
23.41%
66.00%
N1
57.81%
42.19%
34.13%
18.11%
47.76%
N2
77.73%
22.27%
65.25%
10.04%
24.71%
Episode 분석단위
77명에서 199개 국소 episode
C-first
54개 episode
Death-first
83개 episode
우측 중도절단
62개 episode
상태 경사는 적합된 과정 안의 예후정보이지 인과적 치료효과가 아닙니다. N1과 N2는 무작위 배정된 처치가 아닙니다. 해당 상태에 들어가려면 충분히 생존하고 관찰되며 적격 음성을 만들어야 했습니다.
X. 두 잠재 전이 체제는 숨겨져 있던 큰 이질성을 드러냅니다
낮은 진행 체제
P → N1 7.55%
N1 → N2 28.51%
N2 → C 42.07%
C → C 16.46%
관찰 점유율: 68.36%
동일 체제 유지: 97.24%
높은 진행 체제
P → N1 54.34%
N1 → N2 69.77%
N2 → C 79.30%
C → C 61.45%
관찰 점유율: 31.64%
동일 체제 유지: 99.67%
모형
BIC
CV log loss
최소 점유율
수렴
판단
조작적 MM / 1체제
2288.97
0.5068
100.00%
예
주 해석모형
2체제
2250.20
0.4862
31.64%
예
권장 보완모형
3체제
2321.94
0.4835
27.59%
예
더 복잡하고 체제 분리가 약함
4체제
2415.22
0.4822
13.15%
예
한 CV split에서 최저이나 2체제보다 개선폭이 1% 미만
5체제
2530.28
0.4839
5.15%
아니오
선택해서는 안 됨
같은 관찰상태도 미래경로가 크게 다를 수 있습니다
P → N1은 두 체제에서 7.55%와 54.34%로 달랐습니다. P라는 라벨 하나만으로는 상당한 과정 이질성이 가려집니다.
체제는 빠르게 교대하기보다 지속되는 경향이 있습니다
두 체제의 자기전이확률이 모두 97%를 넘었습니다. 매 배양마다 무작위로 바뀌는 것이 아니라 지속적인 전이맥락이 존재할 가능성이 있습니다.
아직 체제에 임상적 이름을 붙일 수는 없습니다
항생제 노출, 기구, 감염원 조절, 호흡기 처치, 상처부담, 균량, 검체 질, 미생물군 교란 등이 가능하지만 현재 자료로 어느 것도 입증되지 않았습니다.
XI. 모형선택은 하나의 절대 승자를 정하는 경기가 아니라 역할분담입니다
임상 질문
우선 모형
이유
근거 경계
3회 음성 protocol은 어떻게 진행되는가?
4상태 조작적 MM
P, N1, N2, C와 양성 재설정을 직접 표현
주 기술·추론 framework
숨은 전이 이질성이 예측을 개선하는가?
2체제 4상태 HMM
AICc, BIC, 예측, 점유율, 분리도의 균형이 가장 좋음
체제는 관찰적이며 임상라벨이 없음
배양 아래에 어떤 잠재 생물학 상태가 있는가?
생물학적 2상태 HMM
일관된 근거경사를 제공
초기상태에 매우 민감하고 탐색적
C와 사망 중 무엇이 먼저 발생하는가?
연속시간 경쟁위험 모형
노출시간과 경쟁 첫 사건을 사용
균질 전이율 근사
말기 궤적은 비선형인가?
GAMM
선형 혼합모형보다 환자수준 CV log loss가 낮음
BIC가 높고 곡선 불확실성이 큼
사망확률이 상태별로 달라야 하는가?
결정적 승자 없음
AICc와 명목 LR은 5상태를, BIC와 Bayes factor는 공통 사망모형을 선호
근거 혼재
Bayesian shrinkage가 예측을 개선하는가?
입증되지 않음
Log-loss 개선구간이 0을 포함하고 Brier 개선은 거의 없음
결론 불충분
단순 pooled 양성·음성 모형으로 충분한가?
아니오
균질성, 무기억성, 예측점검이 실패
참고모형으로만 사용
주 분석
4상태 조작적 MM
보완 분석
2체제 switching HMM
탐색적 생물학
2상태 생물학적 HMM
결과 경로
경쟁위험 모형
말기 곡선
GAMM과 Bayesian 모형
XII. 임상적으로 방어 가능한 주장과 아직 지지되지 않은 주장
현재 방어 가능한 내용
입증되지 않은 내용
첫 적격 음성이 가장 큰 조작적 병목입니다
첫 음성이 생물학적 제거를 뜻합니다
연속 음성이 점차 강한 근거가 됩니다
N1, N2, C가 직접 관찰된 생물학적 상태와 같습니다
세 번째 음성은 의미 있는 확인가치를 유지합니다
음성 2회만으로 모든 상황에서 격리해제가 가능합니다
국소 C 뒤에도 재양성이 검출될 수 있습니다
국소 C가 영구 박멸, PLC, PtLC와 같습니다
추적된 대상에서 검출 재양성이 이른 경우가 많았습니다
모든 제거 환자의 실제 재양성 발생률이 62.96%입니다
마지막 4주의 음성 급증은 지지되지 않았습니다
지속 양성이 임박한 사망을 일으키거나 확실히 예측합니다
현재 상태에 따라 적합된 C 대 사망 경로가 달랐습니다
환자를 N2로 이동시키면 인과적으로 사망률이 낮아집니다
두 잠재 전이 체제가 보완모형을 개선했습니다
코호트가 두 개의 검증된 생물학적 환자형으로 구성됩니다
중요한 보고 공백: 이 export에는 Clinical Bayesian Networks가 없습니다
Section manifest에는 36개 분석 section이 있지만 완료된 Clinical Bayesian Network section은 없습니다. 따라서 이 문서에는 선택된 Bayesian Network 부모변수, 환자 군집 edge stability, BDeu prior sensitivity, 환자분리 network 검증, 기저율 보정 동시집락화 농축도, 새 reservoir-discordance 감사결과가 나타나지 않습니다.
병원체
부위
숙주요인
관찰과정
→
P → N1, N1 → N2, N2 → C, C → P, 사망 또는 고정기간 재양성
XIII. 다음 연구는 아직 해결되지 않은 기전을 직접 겨냥해야 합니다
우선순위 1 · 데이터 고정
50건·19건·30건의 차이를 조정하고 준비 오류 8건을 검토해야 합니다.
우선순위 5 · 가장 느린 저장소
PLC와 PtLC를 maximum-time 또는 weakest-link 결과로 분석해야 합니다.
우선순위 6 · 시간적 동시집락화
이미 알려진 CRE, 같은 날 CRE, trigger 뒤 incident CRE를 분리해야 합니다.
우선순위 7 · 외부검증
이후 SSCH 코호트 또는 독립 LTCF에 고정된 규칙을 적용해야 합니다.
다음 분석
해결하려는 숨은 질문
잠재적 임상가치
전이별 계층모형
각 단계는 어떤 병원체–부위 요인에 의해 결정되는가?
제거 시작기전과 완성기전을 분리
Joint observation–event model
재양성이 생물학적인가, 더 자주 검사되어 검출된 것인가?
합리적인 C 이후 감시일정 설계
체제소속 회귀모형
어떤 임상변수가 두 잠재 체제를 설명하는가?
수정 가능한 전이맥락 탐색
Weakest-link PLC/PtLC 모형
어떤 미해결 저장소가 집계 제거를 붙잡고 있는가?
실제 격리해제를 지연시키는 부위를 표적화
시간적 동시집락화 risk set
다른 MDRO가 미래 CRE 검출을 실제로 농축시키는가?
유효한 screening yield와 number needed to test 계산
시간적 또는 외부검증
파생 코호트 밖에서도 결과가 유지되는가?
전이가능성과 임상 신뢰성 평가
XIV. 최종 임상적 종합
저장소와 병원체 생태
→
P 이탈의 어려움
→
취약한 N1
→
강한 N2 근거
→
국소 C
검출 재양성
↖
숨은 전이 체제
↗
사망 또는 미해결 추적
이 전향적 LTCF 자료에서 MDRO 제거는 단일 전환사건이 아니라 이력 의존적 다단계 과정이었습니다. 가장 큰 장벽은 적격 음성 연속과정을 시작하는 것이었습니다. 음성 근거가 누적될수록 국소 제거 확인이 유리해졌지만 중간상태는 재설정될 수 있었고 국소 C 뒤에도 재양성이 검출될 수 있었습니다. 사망 전 마지막 4주 동안 음성 배양이 증가한다는 근거는 없었습니다. 경쟁위험 모형에서는 미해결 episode가 많았고, 2체제 HMM은 큰 잠재 전이 이질성을 드러냈습니다. 4상태 조작적 MM을 주 분석으로 유지하고 switching HMM, 생물학적 HMM, 말기모형, 경쟁위험 분석은 서로 다른 보완적 역할로 사용해야 합니다.
Written on August 6, 2026
Four complementary models for interpreting intermittent MDRO cultures: an integrated guide (Written August 7, 2026)
Culture surveillance does not observe colonization continuously. It opens a narrow observational window only when a specimen is collected. The biological process may change before, between, and after those culture dates, while the operational clearance rule is constructed afterward from the sequence of observed positive and negative results.
Four complementary analytical layers are therefore needed. The four-state operational Markov model describes the observed clinical rule. The two-regime switching-HMM tests whether one transition matrix is sufficient. The continuous-time latent model reconstructs a probabilistic biological trajectory between irregularly spaced cultures. The Death- and sampling-aware analyses examine whether terminal follow-up and the specimen-collection process distort the apparent culture trajectory.
Central conclusion: these four models are not competing attempts to name one universal winner. They answer four different questions. The four-state MM remains the primary model for the protocol. The two-regime switching-HMM provides supported evidence of heterogeneous transition dynamics. The three-state CT-HMM provides a useful but weakly identified biological interpretation of continuous change between cultures. The duration-aware semi-Markov approximation remains exploratory. The current Death–sampling analysis is a coordinated framework of sensitivity and competing-risk models, not yet one fully unified joint likelihood.
1
Four-state operational MM
What did the clinical three-negative rule actually observe?
2
Two-regime switching-HMM
Is one transition matrix sufficient for every interval?
3
Continuous-time latent process
What may be happening biologically between culture dates?
4
Joint sampling–Death framework
How do terminal truncation and specimen collection affect the result?
I. One biological process, four analytical questions
From continuous biology to intermittent observation
Latent biological process
Colonization burden changes continuously as \(X(t)\)
→
Specimen-collection process
A culture is collected only at selected times \(t_1,t_2,\ldots\)
→
Observed culture result
\(Y(t_k)\) is positive or negative
→
Operational evidence history
P → N1 → N2 → C
Death
can terminate the future opportunity to observe C
Missing or unavailable culture
is not a negative result
How to read the illustration: the continuous curve is conceptual rather than an observed patient record. Culture results are isolated samples from the curve. A negative culture may occur while the latent process is declining, fluctuating near the detection boundary, or already close to sustained clearance.
In a continuous-time latent model, the transition probability across an interval of length \(\Delta t\) is
A 3-day gap and a 30-day gap therefore contribute different transition opportunities. The culture result at a collection date is modeled separately as an imperfect observation:
\[
Y(t_k)\sim p\!\left(y\mid X(t_k)\right).
\]
The four layers answer different questions
Analytical layer
What is directly observed?
What is latent or modeled?
Main question
Principal output
Four-state operational MM
P, N1, N2, and C reconstructed from culture history
Nothing biological is hidden
How does the protocol-defined sequence progress or reset?
Per-next-culture transition probabilities
Two-regime switching-HMM
The same P, N1, N2, and C states
R1 or R2 selects the applicable transition matrix
Is one pooled transition pattern sufficient?
Regime-specific transition phenotypes
Three-state CT-HMM
Positive and negative cultures at exact dates
High Burden, Low or Intermittent Burden, and Sustained Clearance
What continuous latent trajectory is compatible with the intermittent observations?
Transition rates, emissions, and posterior trajectories
Duration-aware phase-type model
The same exact-date cultures
Early and established phases within High and Low Burden
Does time already spent in a latent state matter?
A constrained approximation to semi-Markov duration dependence
Death–sampling framework
Culture dates, culture results, Death, and independent censoring
Death intensity, observation intensity, terminal trajectory, and first-event probabilities
How much do terminal truncation and culture collection affect the apparent process?
Death sensitivity, observation audit, terminal trends, and competing risks
The following domain-coverage map is a conceptual reading aid rather than a formal model-selection statistic. Larger circles indicate that a model addresses more of the named domain.
Best for clinical rule
Four-state operational MM
Best for transition heterogeneity
Two-regime switching-HMM
Best for unobserved interval biology
Three-state CT-HMM
Best for terminal and observation effects
Death–sampling framework
Requires an eligible external endpoint after the sequence timeline
Adjusted terminal trajectory models
Patient-level longitudinal records
60
1,019 collected cultures
Requires Death- or censoring-relative culture windows and covariates
Competing-risk multi-state model
199 pathogen–site episodes
77
54 C-first, 83 Death-first, and 62 right-censored episodes
The unit is one patient × pathogen × site episode
321
Prepared sequences
→
234
Four-state eligible
→
198
HMM and CT-HMM cohort
→
152
Endpoint-linked CT sensitivity
These totals must not be exchanged. A patient may contribute several pathogen–site sequences or episodes, while Death remains a patient-level endpoint.
II. Four-state operational MM: what the protocol actually observed
Operational meaning
P
Positive status
16.78%→
N1
First qualifying negative
55.64%→
N2
Two qualifying negatives
73.10%→
C
Third qualifying negative confirms local clearance
What the state means
How much qualifying negative evidence has accumulated under the protocol.
What the state does not mean
A directly measured organism burden, microbiological eradication, or a validated release threshold.
P, N1, N2, and C are observed evidence-history states. N1 and N2 are not claims about biological burden. They record how much qualifying negative evidence has accumulated under the three-negative rule.
The first negative is the principal bottleneck
Current state
Forward or favorable transition
Competing transition
Interpretation
P
P→N1: 299/1,782 = 16.78%
P→P: 83.22%
Starting a qualifying negative run is difficult.
N1
N1→N2: 153/275 = 55.64%
N1→P: 44.00%
One negative is favorable but remains fragile.
N2
N2→C: 106/145 = 73.10%
N2→P: 26.90%
The third negative retains a strong confirmation role.
C
C→C: 40/69 = 57.97%
C→P: 42.03%
Confirmed local clearance is not necessarily durable.
234
Eligible sequences
→
144
Ever reached N1
61.54%
→
108
Ever reached N2
46.15%
→
88
Ever reached C
37.61%
From all sequences to N1
144/234 = 61.54%
From N1 reachers to N2
108/144 = 75.00%
From N2 reachers to C
88/108 = 81.48%
Forward progression rises from 16.78% to 55.64% and then 73.10%. This is the clearest evidence that the three-negative sequence contains increasing information. The third negative is not a redundant repetition of the first.
The uninterrupted product
\[
0.1678\times0.5564\times0.7310=0.0682
\]
equals 6.82%, but 37.61% of eligible sequences eventually reached C. The product describes one immediate clean run. The eventual fraction also includes persistent positivity, failed runs, resets, and later successful attempts.
Initiation and completion are separate ecological problems
Pathogen–site stratum
P→N1 Initiation
N1→N2 Stabilization
N2→C Completion
C→P Observed recurrence
Operational reading
CRE · Stool
9.50%
48.53%
59.38%
42.86%
Severe initiation bottleneck with additional difficulty later in the run
CRE · Urine
18.75%
57.69%
74.07%
47.06%
Negative runs begin infrequently in absolute terms, but N2 usually completes
VRE · Urine
41.10%
75.00%
95.00%
33.33%
Favorable initiation, stabilization, and completion
MRAB · Sputum
67.86%
63.16%
90.91%
66.67%
Rapid progression, but small counts and uncertain durability
MRPA · Sputum
66.67%
66.67%
60.00%
NE
Runs begin readily, while completion becomes the relative bottleneck
The horizontal axis of the next map represents the ability to start a qualifying negative run. The vertical axis represents the ability to complete C after N2 has already been reached. Bubble size reflects the number of eligible sequences.
Low initiation + lower completion
CRE stool and VRE stool remain difficult across more than one gate.
Low initiation + favorable completion
CRE urine starts negative runs infrequently but often completes after N2.
Easy initiation + weaker completion
MRPA sputum shows why the first negative alone can overstate progress.
Favorable initiation + favorable completion
VRE urine and MRAB sputum occupy the most progressive region.
A stratum can have a low probability of entering N1 but a high probability of completing C once N2 has already been reached. These probabilities are conditional on different source-state populations and should not be expected to move together.
Recurrence requires explicit denominators
Recurrence endpoint
Result
What it means
Transition-level C→P
29/69 = 42.03%
Positive next transition among observed transitions from C
Among followed cleared sequences
21/39 = 53.85%
At least one recurrence where post-C surveillance continued
Among all cleared sequences
21/88 = 23.86%
Includes sequences without a counted post-C transition
Among followed cleared patients
17/27 = 62.96%
At least one recurrent sequence per followed patient
Time to first detected recurrence
Median 11 days; IQR 7–31 days
Detected recurrence was often early
29/69
Transition-level recurrence
21/39
Followed-sequence recurrence
17/27
Followed-patient recurrence
11 days
Median time to first recurrence
The operational MM is therefore the strongest model for explaining the protocol, but it cannot identify the continuous biological state between cultures, distinguish measurement variation from true burden change, or determine why one pathogen–site stratum behaves differently from another.
III. Two-regime switching-HMM: evidence for heterogeneous transition dynamics
What R1 and R2 mean
The switching-HMM preserves the four observed operational states. It adds a hidden regime \(R_n\) that determines which transition matrix applies at observation step \(n\):
R1 and R2 are hidden nodes, but they are transition regimes rather than biological burden states. They are not equivalent to High Burden and Low Burden in the continuous-time model.
The two transition phenotypes are substantially different
Feature
R1
R2
Interpretation
Estimated occupancy
68.36%
31.64%
Most fitted observation intervals were allocated to R1.
P→N1
7.55%
54.34%
Starting a negative run is far easier in R2.
N1→N2
28.51%
69.77%
The first negative is much more stable in R2.
N2→C
42.07%
79.30%
Completion is considerably more likely in R2.
C→C
16.46%
61.45%
Operational C is much less durable in R1.
Regime persistence
R1→R1 97.24%
R2→R2 99.67%
Regime switching is possible but rare per observed transition.
Typical R1 journey
P → P → P → N1 → P
Persistence and reset dominate.
Typical R2 journey
P → N1 → N2 → C
Forward progression dominates.
Meaning of the result: one pooled transition matrix averages together intervals that behave very differently. The data are better represented by a predominant persistent/reset-prone transition mode and a smaller clearance-progressive transition mode.
Why two regimes were selected
Model
Parameters
AICc
BIC
Patient-CV log loss
Model-order reading
Four-state MM
6
2,254.700
2,288.967
0.506774
Directly interpretable baseline
HMM · 2 regimes
15
2,164.659
2,250.204
0.486243
AICc/BIC winner and recommended order
HMM · 3 regimes
26
2,173.920
2,321.938
0.483468
Smaller predictive gain with weaker separation
HMM · 4 regimes
39
2,193.655
2,415.218
0.482243
Single-partition CV winner but heavily penalized
HMM · 5 regimes
54
2,224.257
2,530.284
0.483921
Did not converge
AICc winner
Two hidden regimes
BIC winner
Two hidden regimes
Single CV winner
Four hidden regimes
Defensible recommendation
Two hidden regimes
The two-regime model reduced AICc by about 90 points and BIC by about 39 points relative to the MM, while improving patient-separated log loss by roughly 4%. The four-regime model had a slightly lower single-partition CV loss, but the relative predictive gap from two regimes was only 0.82%, while its BIC was approximately 165 points worse. Two regimes therefore provide the most defensible balance of fit, prediction, occupancy, separation, and parsimony.
What the regimes do not prove
R1 is not automatically “CRE stool,” severe illness, antibiotic exposure, or one fixed patient group.
R2 is not automatically biological clearance or a permanently favorable patient class.
A sequence can move between R1 and R2, although the fitted regimes are highly persistent.
Pathogen, site, treatment, devices, and host condition may help explain regime membership, but that explanatory regression has not yet been completed.
Supported statement
Unsupported overinterpretation
Two transition-dynamics patterns are supported.
Two biological species of patient have been discovered.
R1 is more persistent and reset-prone.
R1 is definitively caused by one pathogen, site, or treatment.
R2 is more clearance-progressive.
R2 proves biological eradication.
Regimes may change over time.
Every sequence belongs permanently to one regime.
The switching-HMM therefore strengthens the conclusion that transition behavior is heterogeneous, but it does not identify the biological cause of that heterogeneity.
IV. Continuous-time latent process and the hidden semi-Markov question
The shortest explanation: reconstructing the movie between snapshots
The four-state MM and switching-HMM analyze the sequence visible at culture dates. A continuous-time latent model asks what biological path may have connected those visible points.
Snapshot analysis
Day 0 positive
Day 7 negative
Day 28 positive
The recorded results are known.
Continuous-time analysis
High Burden may decline toward Low Burden, fluctuate, approach Clearance, and rebound before the next culture.
The unobserved path is inferred probabilistically.
Day 0 Positive
7 unobserved days
Day 7 Negative
21 unobserved days
Day 28 Positive
The observations reveal only three points. They do not reveal whether the latent process changed immediately after day 0, immediately before day 7, several times between days 7 and 28, or not at all. The exact biological transition time is therefore interval-censored.
Essential interpretation: the model estimates a probability distribution over possible trajectories. It does not recover one certain hidden movie or one exact transition date.
What continuous-time, hidden, Markov, and semi-Markov mean
Continuous-time
A latent transition may occur at any time, not only when the next culture is collected. A 3-day gap and a 30-day gap represent different transition opportunities.
Hidden
The true biological state is not observed directly. Only positive or negative culture emissions are observed at selected dates.
Markov
The current latent state determines the future transition rates. Time already spent in that state does not directly change the rate.
Semi-Markov
The probability of leaving a latent state may depend on how long the process has already remained there. Duration becomes part of the model.
In an ordinary CT-HMM, the short-term exit probability is approximately
\[
\Pr(\text{exit during }dt\mid X(t)=i)
\approx q_i\,dt,
\]
where \(q_i\) does not depend directly on the duration already spent in state \(i\). In a hidden semi-Markov model, an elapsed-state-duration variable \(u\) may alter the hazard:
\[
\Pr(\text{exit during }dt\mid X(t)=i,\text{ duration}=u)
\approx h_i(u)\,dt.
\]
The duration chart is conceptual: it illustrates the difference between a duration-independent Markov rate and a duration-dependent semi-Markov rate. It is not a fitted curve from this MDRO dataset.
Markov and semi-Markov are not interchangeable
Feature
Continuous-time HMM
Continuous-time hidden semi-Markov model
Latent state
Hidden
Hidden
Transition timing
Continuous
Continuous
Time already spent in the state
Does not directly affect the exit rate
May directly affect the exit probability
Dwell-time distribution
Usually exponential
Can be non-exponential
Clinical example
A High-Burden state has the same instantaneous exit rate on day 5 and day 100.
A long-established High-Burden state may be harder to leave than a recently entered High-Burden state.
Complexity
Lower
Higher; requires more information about duration
What the current app actually fitted
Important terminology: the current primary result is a Three-State CT-HMM, not a full unrestricted continuous-time hidden semi-Markov model. A separate duration-aware phase-type CT-HMM was fitted as a constrained approximation to semi-Markov behavior.
Model
What it contains
Role in the current analysis
Current status
Three-State CT-HMM
High Burden, Low or Intermittent Burden, and Sustained Clearance with exact culture intervals
Prespecified primary biological model
Selected with caution
Duration-Aware Phase-Type CT-HMM
High and Low Burden are divided into early and established phases
Constrained approximation to semi-Markov duration dependence
Exploratory
Full continuous-time HSMM
Explicit and flexible dwell-time distributions for each latent state
Not fitted in the current implementation
Not yet implemented
H1
High · early
→
H2
High · established
→
L1
Low · early
→
L2
Low · established
→
C
Sustained clearance
L1 or L2 may rebound toward H1
C may return toward L1
The phase expansion gives the model some memory of state duration. A process in H2 has, by construction, remained in High Burden longer than a process in H1. This is a practical semi-Markov approximation, but it does not estimate an unrestricted dwell-time curve.
The current primary model is a three-state CT-HMM
High Burden
Culture-positive probability 98.88%
⇄
Low or Intermittent Burden
Culture-positive probability 83.57%
⇄
Sustained Clearance
Culture-positive probability 13.02%
Latent state
Positive emission
Negative emission
Practical reading
High Burden
98.88%
1.12%
Almost always appears positive.
Low or Intermittent Burden
83.57%
16.43%
May occasionally appear negative despite continued latent burden.
Sustained Clearance
13.02%
86.98%
Usually appears negative, but the fitted state is not perfectly absorbing or perfectly observed.
These are probabilistic latent states. “Low or Intermittent Burden” is not equivalent to a negative culture: the fitted model still assigns an 83.57% positive-emission probability to that state. Likewise, “Sustained Clearance” is not a perfect absorbing state because a positive emission remains possible and the model permits return to Low Burden.
Model comparison supports three states, but not an unrestricted semi-Markov model
Candidate
Parameters
AICc
BIC
Patient-CV log loss
Optimization interpretation
Two-State CT-HMM
4
2,152.085
2,174.937
0.479704
Objective-stable, but strict parameter convergence not reached
Three-State CT-HMM
7
2,134.926
2,174.898
0.476245
Prespecified and criterion-selected; flat likelihood ridge remains
Duration-Aware Phase-Type CT-HMM
12
2,145.026
2,213.495
0.475017
Small CV advantage, but objective stability was not reached
The three-state model improves AICc by 17.16 points relative to the two-state model. Their BIC values differ by only 0.04 points, which is effectively a tie. The duration-aware model improves CV log loss over the three-state model by only 0.00123, approximately 0.26%, while worsening BIC by 38.60 points.
The data therefore support retaining an intermediate Low or Intermittent Burden state for interpretation, but they do not prove that exactly three biological states exist. More importantly, the duration-aware extension does not receive enough support to replace the simpler three-state CT-HMM.
Direct answer: the dataset supports continuous-time latent modeling with caution. It does not currently support the stronger statement that a full hidden semi-Markov duration model is required.
Initiation, rebound, completion, and recurrence become continuous-time rates
Latent transition
Rate per 100 days
Single-edge expected waiting time
Biological interpretation
High → Low
1.0056
99.4 days
Initiation of a reduction from high burden
Low → High
0.6452
155.0 days
Rebound toward high detectability
Low → Clearance
2.8338
35.3 days
Completion into sustained clearance
Clearance → Low
2.8001
35.7 days
Latent recurrence or renewed intermittent detectability
High Burden
1.0056 / 100 dinitiation →← rebound 0.6452
Low or Intermittent Burden
2.8338 / 100 dcompletion →← recurrence 2.8001
Sustained Clearance
The slowest principal step is High→Low initiation. Once the process is in Low Burden, the completion rate exceeds the rebound rate.
Under a constant-rate competing-exit interpretation, the conditional probability that the next exit from Low Burden goes to Sustained Clearance is
These are model-derived calculations, not directly observed patient probabilities. They nonetheless reinforce the same stage-specific logic seen in the operational model: entering a lower-burden process and completing sustained clearance are different events.
Pathogen–site continuous-time domain map
The following map places the regularized High→Low initiation rate ratio on the horizontal axis and the Low→Clearance completion rate ratio on the vertical axis. A value above 1 indicates a faster rate than the pooled three-state model. Bubble size approximates the number of included sequences. Sparse cells are omitted from the principal map.
Stratum
Sequences
Initiation RR
Completion RR
Rebound RR
Recurrence RR
Mean High
Mean Clearance
CRE · Stool
60
0.880
0.695
2.209
1.265
54.48%
11.55%
CRE · Sputum
23
0.927
1.054
1.307
1.681
37.24%
19.59%
CRE · Urine
29
1.408
0.843
1.101
0.705
36.75%
24.34%
VRE · Stool
33
0.922
0.903
0.973
1.297
41.57%
17.87%
VRE · Urine
21
1.746
2.043
0.625
0.574
12.22%
50.60%
MRAB · Sputum
11
1.926
2.937
0.470
0.773
1.90%
58.21%
CRE stool and VRE urine are the most internally consistent signals across the operational and continuous-time layers. CRE stool has slow initiation, slow completion, strong rebound, and high posterior High Burden. VRE urine has rapid initiation and completion, weaker rebound, and high posterior Sustained Clearance.
CRE urine demonstrates why the models must remain separate. Its operational P→N1 probability is low in absolute terms, but its latent High→Low rate is faster than the pooled rate. At the same time, its Low→Clearance rate is below the pooled rate. A plausible model-based reading is that CRE urine can leave the high-burden state without immediately producing a stable sequence of observed negatives and may remain in an intermittently detectable state before full completion.
Does the current result support model 3?
Proposed conclusion
Current evidence
Verdict
Unequal culture intervals should not be treated as equal steps.
Exact calendar intervals were used for 2,445 cultures from 198 sequences and 79 patients.
Supported
An intermediate Low or Intermittent Burden state is useful.
The three-state model improved AICc by 17.16 points over the two-state model and had slightly better patient-CV prediction.
Supported with caution
The three-state structure is clearly superior by every criterion.
The two-state and three-state BIC values differ by only 0.04 points.
Not decisively established
Duration dependence requires a semi-Markov model.
The duration-aware candidate improved CV log loss by only 0.00123, while BIC worsened by 38.60 points.
Not supported as the primary model
The exact latent transition time is known.
Transitions remain interval-censored between cultures.
Not supported
The latent states are measured organism loads.
The states are inferred from binary culture emissions without a quantitative microbiological gold standard.
Not supported
Verdict for model 3: the dataset supports a continuous-time latent interpretation with caution. It supports separating initiation from completion and retaining an intermediate Low or Intermittent Burden state as a useful explanation. It does not currently support the stronger claim that a full hidden semi-Markov duration model is required.
Identification and convergence boundary
The three-state likelihood became objective-stable, but no start met the strict parameter-simplex convergence criterion.
The likelihood surface appears relatively flat, so several nearby rate combinations may explain the culture sequence similarly well.
Emission probabilities can absorb sampling variation, assay behavior, unmeasured treatment, and pathogen–site heterogeneity.
The pathogen–site rate ratios are regularized toward the pooled model and are not a fully estimated Bayesian hierarchy.
The duration-aware candidate did not reach objective stability and therefore remains exploratory.
Relatively stable
Overall likelihood level and broad High–Low–Clearance interpretation
Less stable
Exact placement of individual transition rates on a flat likelihood ridge
Not directly observed
True quantitative organism burden, exact transition time, and unrestricted dwell-time distribution
The continuous-time model is therefore best described as a supported-with-caution latent process interpretation, not as a direct measurement of biological burden.
V. Joint sampling–Death framework: what it means and what is currently supported
The shortest explanation: analyzing both the movie and the camera
A culture record is produced only when a specimen is successfully collected. Death then permanently ends future culture opportunities. The observed culture history is therefore produced by both a biological process and an observation process.
Biological process
Colonization burden changes continuously whether or not a specimen is collected.
Sampling process
Culture timing may depend on the surveillance schedule, specimen availability, clinical condition, devices, or earlier results.
Observed result
Positive or negative is observed only at a successful collection date.
Death process
Death permanently ends future opportunities to collect cultures and confirm C.
Purpose of including Death: the objective is not merely to ask whether negative cultures increase before Death. The analysis also asks whether negative cultures decrease, whether sustained-clearance probability falls, whether biological transition rates change after Death is modeled, and whether Death removes the future opportunity to complete C.
Three observation distortions must be separated
Distortion
Example
Why ordinary culture analysis can be misleading
Informative sampling
Cultures are collected more or less frequently in certain latent states.
The observed culture series overrepresents states with more collection opportunities.
Specimen unavailability
A sputum, urine, or stool specimen cannot be obtained.
No culture result is produced. The missing result must not be converted into a negative result.
Terminal truncation by Death
A patient dies before a third qualifying negative can be collected.
The episode can never subsequently be observed to reach C, even if the latent process had begun to improve.
Positive
→
First negative
→
No obtainable specimen
→
Death
This sequence does not contain a second negative or a third negative. It contains one negative, one missing observation opportunity, and a terminal endpoint.
What a fully joint model would contain
Latent state \(X(t)\)
High, Low, or Clearance
→
Sampling intensity \(\lambda_N(t)\)
Whether and when a specimen is collected
→
Culture \(Y(t_k)\)
Positive or negative emission
↓
Death intensity \(\lambda_D(t)\)
Death terminates future sampling and culture observation
A fully joint likelihood would estimate these processes together:
In this expression, \(X\) is the unobserved biological trajectory, \(N\) is the specimen-collection process, \(Y\) is the observed culture result, and \(D\) is the Death process.
What the current app actually fitted
Important terminology: the current implementation is not yet one fully unified joint sampling–Death likelihood. It is a coordinated framework of several linked analyses.
Culture-only CT-HMM
+
Death-aware CT sensitivity
+
Observation-time audit
+
Reverse-time terminal models
+
C-versus-Death competing risk
Current component
Question answered
What it does not answer alone
Death-aware CT-HMM sensitivity
Do biological transition rates move when Death is incorporated?
It does not prove that latent burden causes Death.
Observation-time audit
Do fitted culture intensities differ across latent states?
It does not establish the literal clinical sampling mechanism.
Reverse-time terminal models
Do observed negativity and latent-state probabilities change near Death?
They do not identify a causal terminal-illness mechanism.
Competing-risk multi-state model
Does confirmed C or Death occur first?
It excludes recurrence after C and does not model the full latent burden trajectory.
Patient-balanced composite likelihood is used in the Death-aware sensitivity so that one patient contributing several pathogen–site sequences does not contribute several full copies of the same patient-level Death endpoint.
Death raises three distinct analytical questions
Terminal trajectory
Do observed negatives or latent-clearance probability rise or fall as Death approaches?
Transition robustness
Do High→Low, Low→High, Low→C, and C→Low rates change when Death is included?
First-event competition
From P, N1, or N2, does confirmed C or Death occur first?
These questions are related but not interchangeable. A favorable C-before-Death probability at N2 does not prove that negative cultures increase near Death. A terminal decline in negative cultures does not by itself reveal which operational or latent gate deteriorated.
What each current component contributes
Component
Question
Current result
Evidence boundary
Death-aware CT-HMM sensitivity
Do latent biological rates change when Death is added?
Shared Death intensity retained; biological rates moved by approximately 7%–13%
Associational and based on the endpoint-linked composite cohort
Observation-time audit
Does culture timing vary by latent state?
Exported narrative and numerical criteria conflict
Current conclusion must remain unresolved
Reverse-time terminal analysis
Do observed negatives or latent clearance change near Death?
No terminal negative surge; sustained-clearance probability falls
Observational and conditional on collected cultures
Competing-risk multi-state model
Does confirmed C or Death occur first?
C-before-Death probability rises from P to N1 to N2
First-event model; post-C recurrence is excluded
Adding Death moderately changes rates but not the overall architecture
Death structure
AICc
BIC
Interpretation
Shared Death intensity
951.119
986.807
Preferred within the directly comparable Death-structure pair
State-specific Death intensities
952.952
997.500
Additional state-specific hazards were not justified
Biological edge
Relative change after adding Death
Interpretation
High→Low
−13.28%
Moderate reduction in fitted initiation rate
Low→High
−10.31%
Moderate reduction in fitted rebound rate
Low→Clearance
−7.13%
Completion estimate remains broadly similar
Clearance→Low
−7.04%
Recurrence-like dynamics remain broadly similar
Death is therefore not irrelevant to the fitted colonization process. The rate estimates move after Death is incorporated. However, Death does not overturn the basic High–Low–Clearance architecture.
Meaning of the shared-intensity result: the result does not show that Death is unrelated to colonization state. It shows only that the current data do not justify estimating a separate Death intensity for every latent biological state.
The absolute culture-only and Death-aware rates in this sensitivity branch use a restricted endpoint-linked composite cohort. Their relative changes are more interpretable than direct comparison with the full-cohort primary rates.
Terminal change involves fewer negatives and lower sustained-clearance probability
Terminal comparison
Death cohort
Independent-censoring cohort
Interpretation
Observed negative fraction
Terminal minus earlier: −8.53 pp; 95% interval −14.08 to −2.69
−2.05 pp; interval −15.07 to +9.53
No supported terminal negative surge before Death
Latent High Burden
+0.25 pp
−0.01 pp
High burden is nearly unchanged in complete windows
Latent Sustained Clearance
−3.23 pp
+3.19 pp
Clearance probability moves in opposite directions
Inferred Low/Intermittent Burden shift
Approximately +2.98 pp
Approximately −3.18 pp
Arithmetic decomposition of the three posterior states
Earlier sustained clearance 21.57%
→
Death-terminal sustained clearance 18.34%
+
Posterior mass shifts mainly toward Low/Intermittent Burden
The model-derived terminal pattern is more consistent with failure to complete or maintain sustained clearance than with a sudden surge into High Burden. This remains an inference conditional on the fitted CT-HMM and does not establish that terminal illness caused the shift.
The Bayesian observed-culture model reached a similar directional conclusion: Death-terminal minus earlier negativity was −2.24 percentage points, with a 95% credible interval from −8.19 to +3.58, and only a 22.85% posterior probability of an increase. The Death-aligned change was also substantially less favorable than the censoring-aligned change.
Direct answer to the Death question: accounting for Death did not reveal an excess of negative cultures before Death. The more coherent finding was fewer observed negatives and a lower probability of sustained clearance.
Competing risk measures the remaining opportunity to complete C
Starting state
Eventual C before Death
Death before C
90-day C
90-day Death
Still P/N1/N2 at 90 days
P
42.07%
57.93%
10.59%
23.41%
66.00%
N1
57.81%
42.19%
34.13%
18.11%
47.76%
N2
77.73%
22.27%
65.25%
10.04%
24.71%
P
Long path remains
→
N1
One negative accumulated
→
N2
Only one qualifying negative remains
→
C-before-Death rises
42.07% → 57.81% → 77.73%
N2 has a high C-before-Death probability because only one additional qualifying negative is required. This does not mean that negative cultures causally prevent Death. It means that an episode already close to the operational endpoint has a shorter remaining pathway to C.
The observation-process result is currently unresolved
Publication-critical audit: the exported narrative states that a shared observation intensity was preferred and that state-dependent culture timing lacked BIC support. However, the displayed numerical criteria are substantially lower for the state-specific model. This internal inconsistency must be resolved before the observation-process result is used in a scientific conclusion.
Observation-time fit
AICc
BIC
Numerical reading
Shared intensity
7,342.259
7,380.883
Higher criterion values
State-specific intensities
6,448.795
6,497.031
Lower by 893.46 AICc and 883.85 BIC points
Latent state
Fitted observation intensity per day
Expected days between cultures
Audit concern
High Burden
0.13086
7.64 days
Compatible with approximately weekly surveillance
Low or Intermittent Burden
0.00106
941.02 days
Implausible under the scheduled surveillance design
Sustained Clearance
0.12088
8.27 days
Compatible with approximately weekly surveillance
The extreme Low-Burden observation interval suggests weak identification or model misspecification under the Poisson observation assumption. The 941-day estimate should not be treated as a literal clinical sampling interval.
The study used approximately weekly scheduled surveillance. The Poisson observation-time model is therefore a sensitivity analysis rather than a literal description of how clinicians ordered cultures.
Until the selection logic and reported criteria are reconciled, the defensible conclusion is that the influence of informative culture timing remains unresolved.
Does the current result support model 4?
Proposed conclusion
Current evidence
Verdict
Death should be considered when interpreting the culture trajectory.
Adding Death moved biological rates by approximately 7%–13%, and Death competes with future clearance confirmation.
Supported with caution
Each latent state requires a different Death intensity.
Shared Death intensity had lower BIC than state-specific Death intensities.
Not supported
Negative cultures surge before Death.
Observed and adjusted terminal analyses did not support an increase.
Hypothesis not supported
Negative cultures may become less frequent near Death.
The paired terminal-minus-earlier negative fraction was −8.53 percentage points.
Supported descriptively
Sustained-clearance probability may decline near Death.
The complete-window latent contrast was −3.23 percentage points.
Supported with caution
Death reduces the future opportunity to complete C.
The competing-risk model shows substantial Death-before-C probability, especially from P.
Supported with caution
Culture timing has been successfully separated from latent state.
The observation-process narrative, information criteria, and fitted Low-state interval conflict.
Unresolved
A fully joint sampling–Death likelihood has been fitted.
The current app fits linked components rather than one unified likelihood.
Not yet implemented
Verdict for model 4: the current results support considering Death as a competing and observation-terminating process. They do not support a state-specific Death hazard, and they do not yet establish that the sampling process has been successfully separated. The phrase “joint sampling–Death model” therefore describes the intended analytical framework more accurately than the present single fitted model.
VI. The same culture sequence viewed through four lenses
Worked example
Day 0 Positive
→
Day 7 Positive
→
Day 14 Negative
→
Day 21 Negative
→
Day 28 Positive
→
Day 35 No further culture
Four-state MM
P → P → N1 → N2 → P
The negative run reached N2 but reset before C.
Switching-HMM
R1 or R2 governs each operational transition.
The interval may shift from progressive to reset-prone dynamics.
Three-state CT-HMM
High → Low → possible rebound toward High
The negative pair need not equal instantaneous biological clearance.
Death–sampling framework
Later observation opportunity may be truncated.
No culture is not the same as a negative culture.
Model
Interpretation of the same sequence
What is learned
What remains unknown
Four-state MM
P→P→N1→N2→P
Two negatives accumulated, but the run reset before C.
The biological burden between dates
Two-regime switching-HMM
The transition pattern may be allocated to R1, R2, or a switch between them.
Whether this interval resembles a persistent or progressive transition mode
Why the regime occurred
Three-state CT-HMM
Posterior probability may move High→Low during the negative pair and return toward High after the positive result.
A continuous probabilistic trajectory between cultures
Direct quantitative burden and exact transition times
Duration-aware approximation
The interpretation may differ depending on whether High or Low Burden is newly entered or long established.
A constrained test of duration dependence
An unrestricted dwell-time distribution
Death–sampling framework
The absence of a later culture may reflect follow-up termination, specimen availability, discharge, or Death.
How much future clearance opportunity was truncated
The unobserved culture result that would have occurred later
VII. Integrated interpretation of the current dataset
Conclusions that are consistent across model layers
Question
Integrated answer
Supporting layers
Confidence boundary
Where is the main clearance bottleneck?
At the beginning. P→N1 is only 16.78%, and latent High→Low initiation is slow.
Operational MM and CT-HMM
Strong for the observed process; biological mechanism remains latent
Does negative evidence strengthen progressively?
Yes. N1 is fragile, N2 is much more favorable, and the third negative remains meaningful.
Operational MM and competing risk
Strong for the protocol-defined evidence sequence
Is one transition matrix sufficient?
No. Two hidden transition regimes materially improve fit and prediction.
Switching-HMM
Supported, but regime causes remain unknown
Is clearance an instantaneous binary switch?
Probably not. An intermediate low/intermittent state improves the model and explains isolated negatives.
Three-state CT-HMM
Supported with caution because of weak identification
Is a full semi-Markov model required?
Not by the current evidence. The duration-aware candidate adds little prediction and a large BIC penalty.
Duration-aware model comparison
Exploratory only
Is confirmed local clearance durable?
Not necessarily. Operational recurrence and latent Clearance→Low dynamics are both substantial.
Operational recurrence and CT-HMM
Follow-up dependent
Do negatives surge before Death?
No supported surge. Observed negativity is generally lower, and latent sustained clearance declines.
Terminal, Bayesian, and CT reverse-time analyses
Observational, non-causal
Does Death require state-specific latent hazards?
Not in the current CT sensitivity. A shared Death intensity was preferred.
Death-aware CT-HMM
Endpoint-linked composite cohort only
Is sampling state-dependent?
Unresolved. The current narrative and numerical criteria conflict.
Observation-process audit
Must be corrected before scientific interpretation
Has one fully joint sampling–Death model been fitted?
No. The current result combines linked analyses.
Implementation audit
Future methodological extension
Integrated signal
Four-state MM
Switching-HMM
CT-HMM / semi-Markov
Death–sampling framework
Initiation is difficult
Direct support
Regime-dependent support
High→Low is slow
Secondary
Later negative evidence is stronger
Direct support
Especially in R2
Compatible
C-before-Death rises
Clearance is reversible
C→P observed
C durability differs by regime
C→Low modeled
Post-C recurrence outside first-event model
Duration dependence
Not modeled
Not modeled
Exploratory phase-type test
Not primary
Terminal negative surge
Not designed for this question
Not designed for this question
Clearance posterior falls
No supported surge
Sampling-process distortion
Not modeled
Not modeled
Sensitivity audit only
Unresolved audit discrepancy
Pathogen–site ecological synthesis
CRE stool is the most consistent persistence signal. It rarely starts a negative run, has slow latent initiation and completion, rebounds strongly, and spends the largest share of time in High Burden.
VRE urine is the most consistently favorable common stratum. Operational initiation and completion are favorable, latent initiation and completion are rapid, and rebound and recurrence rate ratios are below the pooled model.
Stool overall has slower initiation and completion, stronger rebound, and a lower latent-clearance share than urine.
Urine overall has faster initiation and completion, weaker rebound, and a higher latent-clearance share.
MRAB and MRPA strata often show favorable conversion, but several cells remain small or regularized and should not be treated as precise biological constants.
CRE urine demonstrates that an observed first negative and a latent High→Low transition are not the same event. It may leave High Burden relatively quickly while remaining intermittently detectable before sustained clearance.
CRE stool
Slow start
Slow completion
Strong rebound
High posterior burden
VRE urine
Faster start
Faster completion
Weaker rebound
High posterior clearance
CRE urine
Operational N1 is uncommon
Latent High→Low is faster
Full completion remains slower
MRPA sputum
Negative runs start readily
Completion is the relative bottleneck
VIII. Defensible reporting hierarchy
Recommended model roles
Primary clinical interpretation
Four-state operational MM
Terminal and observation context
Death-aware, sampling, and competing-risk analyses
Reporting position
Model
Recommended claim
Claim to avoid
Primary
Four-state operational MM
The first qualifying negative is the principal bottleneck, and the third negative remains a meaningful confirmation step.
P, N1, N2, and C are biological states.
Supported complementary
Two-regime switching-HMM
Transition dynamics are heterogeneous and are better represented by persistent and progressive latent regimes.
R1 and R2 are validated patient or pathogen subtypes.
Biological-process extension
Three-state CT-HMM
An intermediate low/intermittent state provides a useful continuous-time explanation of cultures between sampling dates.
The fitted latent states are directly measured organism loads.
Exploratory duration sensitivity
Phase-type semi-Markov approximation
Duration dependence remains plausible but is not supported strongly enough to replace the three-state CT-HMM.
A full hidden semi-Markov model has been conclusively selected.
Terminal and prognostic context
Death-aware CT sensitivity, terminal models, and competing risk
Death truncates clearance opportunity, no terminal negative surge is supported, and C-before-Death probability rises with accumulated negative evidence.
Negative cultures causally prevent Death.
Pending audit
Observation-process model
The effect of informative culture timing remains unresolved.
Shared or state-specific sampling has been definitively established.
Future implementation
Unified joint sampling–Death likelihood
A future model should estimate biological state, sampling, culture emission, and Death together.
The current linked framework is already one completed joint likelihood.
Final synthesis
The four-state MM provides the clearest clinical message: most positive sequences remain positive, one negative is fragile, two negatives are strong but incomplete evidence, and the third negative retains a genuine confirmation role. Post-clearance recurrence prevents local C from being interpreted as permanent eradication.
The two-regime switching-HMM shows that the pooled transition matrix conceals two markedly different dynamic modes. Most fitted intervals belong to a persistent, reset-prone regime, while a smaller portion belongs to a clearance-progressive regime. These are transition phenotypes, not validated biological classes.
The three-state CT-HMM adds the most useful continuous interpretation. The principal latent bottleneck is leaving High Burden. Once Low or Intermittent Burden is reached, completion into Sustained Clearance is more likely than rebound, but Low Burden remains frequently culture-positive and Sustained Clearance remains reversible. The model is biologically informative but weakly identified and should remain secondary to the operational result.
The duration-aware phase-type model asks whether recently entered and long-established latent states behave differently. The current predictive gain is very small, its BIC is substantially worse, and optimization is less stable. It therefore does not justify describing the present analysis as a fully supported hidden semi-Markov model.
Death-aware analyses indicate that terminal follow-up does not create an artificial surge of negative cultures. The more coherent pattern is a reduction in observed negativity and sustained-clearance probability, with posterior mass shifting toward low/intermittent burden rather than a major increase in high burden. Accumulated negative evidence nevertheless improves the conditional probability of reaching C before Death because less of the clearance pathway remains.
The sampling component remains unresolved. The displayed observation-process information criteria and the exported narrative conflict, and the fitted Low-state observation interval is not clinically plausible under approximately weekly surveillance. A fully unified joint sampling–Death likelihood has not yet been fitted.
Observed rule
P → N1 → N2 → C
Hidden transition mode
R1 ↔ R2
Latent biology
High ↔ Low ↔ Clearance
Duration question
Early versus established phases
Terminal context
Sampling + Death + unresolved follow-up
Most defensible overall statement: intermittent MDRO cultures reveal a staged and heterogeneous clearance process. The observed protocol is best described by the four-state MM, transition heterogeneity by a two-regime switching-HMM, and the unobserved interval process by a three-state CT-HMM. Full semi-Markov duration dependence is not currently supported as the primary structure. Death truncates the opportunity for confirmation and is associated with reduced negativity and sustained-clearance probability near the terminal endpoint. The contribution of informative sampling remains unresolved pending correction of the observation-process audit.
IX. Final explanation for a first-time reader
Model 3 in one paragraph
Model 3 treats the biological state as a continuous, hidden process and the culture results as occasional observations of that process. The current dataset supports the Three-State CT-HMM as a useful secondary interpretation: the main latent bottleneck is leaving High Burden, and an intermediate Low or Intermittent Burden state helps explain why one negative culture need not equal biological clearance. However, strict convergence and identifiability limitations remain, and the stronger semi-Markov claim that duration itself must alter transition behavior is not supported by the current model comparison.
Model 4 in one paragraph
Model 4 asks whether the observed culture history is partly shaped by when specimens were collected and by Death ending future observation. The Death-related components are informative: Death moderately changes fitted rates, terminal negativity does not increase, sustained-clearance probability falls near Death, and Death competes with C before the clearance sequence can be completed. The sampling component remains unresolved because the current observation-process output is internally inconsistent and produces an implausible Low-state sampling interval. A fully unified joint sampling–Death model has therefore not yet been established.
Most defensible two-line conclusion
Continuous-time conclusion
Supported with caution as a Three-State CT-HMM interpretation. Full semi-Markov duration dependence remains exploratory and is not currently supported as the primary structure.
Sampling–Death conclusion
Death-aware sensitivity and competing risk are supported with caution. Sampling-process separation is unresolved, and one fully joint model has not yet been fitted.
간헐적 MDRO 배양을 해석하는 네 가지 상호보완적 모델: 통합 안내서
배양 감시는 집락화 상태를 연속적으로 관찰하지 못한다. 검체가 채취된 순간에만 좁은 관찰 창이 열린다. 실제 생물학적 과정은 배양 전, 배양 사이, 마지막 배양 이후에도 계속 변할 수 있으며, operational clearance rule은 관찰된 양성·음성 결과의 순서를 이용하여 사후적으로 구성된다.
따라서 네 개의 상호보완적 분석 층이 필요하다. 4-state operational Markov model은 관찰된 임상 규칙을 설명한다. 2-regime switching-HMM은 하나의 transition matrix만으로 충분한지를 검정한다. Continuous-time latent model은 불규칙한 배양 사이에서 가능한 잠재 생물학적 trajectory를 확률적으로 복원한다. Death 및 sampling-aware 분석은 terminal follow-up과 검체 채취 과정이 관찰된 culture trajectory를 왜곡하는지를 평가한다.
핵심 결론: 네 모델은 하나의 universal winner를 결정하기 위한 경쟁 모델이 아니다. 서로 다른 네 질문을 답한다. 4-state MM은 protocol 해석의 primary model이다. 2-regime switching-HMM은 transition dynamics가 heterogeneous하다는 supported evidence를 제공한다. Three-state CT-HMM은 배양 사이의 continuous change에 대한 유용하지만 weakly identified된 biological interpretation을 제공한다. Duration-aware semi-Markov approximation은 exploratory 수준에 머문다. 현재 Death–sampling 분석은 여러 sensitivity 및 competing-risk model을 연결한 framework이며, 아직 하나의 완전한 joint likelihood는 아니다.
1
4-state operational MM
Three-negative clinical rule에서 실제로 무엇이 관찰되었는가?
2
2-regime switching-HMM
모든 interval에 하나의 transition matrix로 충분한가?
3
Continuous-time latent process
배양 날짜 사이에서 생물학적으로 무엇이 일어날 수 있는가?
4
Joint sampling–Death framework
Terminal truncation과 검체 채취가 결과에 어떤 영향을 주는가?
I. 하나의 생물학적 과정과 네 가지 분석 질문
연속적인 생물학에서 간헐적인 관찰까지
잠재 생물학적 과정
집락화 부담이 \(X(t)\)로 연속적으로 변함
→
검체 채취 과정
선택된 시점 \(t_1,t_2,\ldots\)에서만 배양 시행
→
관찰된 배양 결과
\(Y(t_k)\)는 positive 또는 negative
→
Operational evidence history
P → N1 → N2 → C
Death
이후 C를 관찰할 기회를 종료시킬 수 있음
Missing 또는 unavailable culture
음성 결과가 아님
그림을 읽는 방법: continuous curve는 실제 한 환자의 측정값이 아니라 개념도이다. Culture result는 curve에서 간헐적으로 얻은 sample이다. Negative culture는 latent process가 감소 중이거나, detection boundary 근처에서 변동하거나, sustained clearance에 가까워진 상태에서 나타날 수 있다.
Continuous-time latent model에서 길이 \(\Delta t\)인 구간의 transition probability는 다음과 같다.
Death intensity, observation intensity, terminal trajectory 및 first-event probability
Terminal truncation과 culture collection이 apparent process에 어떤 영향을 주는가?
Death sensitivity, observation audit, terminal trend 및 competing risk
다음 domain-coverage map은 formal model-selection statistic이 아니라 이해를 돕기 위한 개념도이다. Circle이 클수록 해당 model이 그 domain을 더 직접적으로 다룬다.
임상 규칙 설명에 가장 적합
4-state operational MM
Transition heterogeneity에 가장 적합
2-regime switching-HMM
관찰되지 않은 interval biology에 가장 적합
Three-state CT-HMM
Terminal 및 observation effect에 가장 적합
Death–sampling framework
Cohort와 denominator를 분리해야 하는 이유
분석
Sequences 또는 episodes
Patients
Observations 또는 transitions
Denominator가 다른 이유
4-state operational MM
234 sequences
91
2,505 operational observations
Observed 또는 documented positive evidence가 필요함
Switching-HMM comparison
198 sequences
79
2,445 observations; 2,247 scored transitions
최소 3개의 operational observation이 필요함
Culture-only continuous-time latent model
198 sequences
79
2,445 cultures
Observed-positive continuous-time trajectory를 사용함
Death-aware CT-HMM sensitivity
152 endpoint-linked sequences
63
Death 35명, independent censoring 28명
Sequence timeline 이후의 적격 외부 endpoint가 필요함
Adjusted terminal trajectory models
Patient-level longitudinal records
60
수집된 culture 1,019건
Death 또는 censoring-relative culture window와 covariate가 필요함
Competing-risk multi-state model
199 pathogen–site episodes
77
C-first 54, Death-first 83, right-censored 62
분석 단위가 patient × pathogen × site episode임
321
Prepared sequences
→
234
Four-state eligible
→
198
HMM 및 CT-HMM cohort
→
152
Endpoint-linked CT sensitivity
이 수치들은 서로 바꾸어 사용할 수 없다. 한 환자는 여러 pathogen–site sequence 또는 episode를 제공할 수 있지만 Death는 patient-level endpoint이다.
II. 4-state operational MM: protocol에서 실제로 관찰한 것
Operational 의미
P
Positive status
16.78%→
N1
첫 qualifying negative
55.64%→
N2
두 번의 qualifying negative
73.10%→
C
세 번째 qualifying negative로 local clearance 확인
State가 의미하는 것
Protocol에 따라 qualifying negative evidence가 얼마나 누적되었는가.
State가 의미하지 않는 것
직접 측정된 organism burden, microbiological eradication 또는 검증된 release threshold.
P, N1, N2, C는 관찰된 evidence-history state이다. N1과 N2는 biological burden을 의미하지 않는다. Three-negative rule에 따라 qualifying negative evidence가 얼마나 누적되었는지를 기록한다.
첫 음성이 가장 큰 병목이다
현재 state
Forward 또는 favorable transition
경쟁 transition
해석
P
P→N1: 299/1,782 = 16.78%
P→P: 83.22%
Qualifying negative run을 시작하기 어렵다.
N1
N1→N2: 153/275 = 55.64%
N1→P: 44.00%
한 번의 음성은 유리하지만 여전히 fragile하다.
N2
N2→C: 106/145 = 73.10%
N2→P: 26.90%
세 번째 음성이 강한 confirmation 역할을 유지한다.
C
C→C: 40/69 = 57.97%
C→P: 42.03%
Confirmed local clearance가 반드시 durable한 것은 아니다.
234
Eligible sequences
→
144
한 번이라도 N1 도달
61.54%
→
108
한 번이라도 N2 도달
46.15%
→
88
한 번이라도 C 도달
37.61%
전체 sequence에서 N1까지
144/234 = 61.54%
N1 도달자에서 N2까지
108/144 = 75.00%
N2 도달자에서 C까지
88/108 = 81.48%
Forward progression은 16.78%에서 55.64%, 다시 73.10%로 상승한다. 이는 three-negative sequence가 점차 강해지는 정보를 포함한다는 가장 명확한 근거이다. 세 번째 음성은 첫 음성을 단순히 반복하는 불필요한 검사가 아니다.
중단 없는 product는
\[
0.1678\times0.5564\times0.7310=0.0682
\]
로 6.82%이지만, eligible sequence의 37.61%는 관찰 기간 중 결국 C에 도달했다. Product는 한 번의 즉시 성공 run을 나타내고, eventual fraction은 지속 양성, 실패한 run, reset, 이후의 재시도와 성공을 포함한다.
시작과 완성은 서로 다른 생태학적 문제이다
Pathogen–site stratum
P→N1 시작
N1→N2 안정화
N2→C 완성
C→P 관찰 recurrence
Operational 해석
CRE · Stool
9.50%
48.53%
59.38%
42.86%
심한 시작 병목에 이후 단계의 어려움도 동반됨
CRE · Urine
18.75%
57.69%
74.07%
47.06%
절대적으로 음성 run은 드물게 시작되지만 N2는 대체로 완성됨
VRE · Urine
41.10%
75.00%
95.00%
33.33%
시작, 안정화, 완성이 모두 유리함
MRAB · Sputum
67.86%
63.16%
90.91%
66.67%
빠른 progression이나 표본이 작고 durability가 불확실함
MRPA · Sputum
66.67%
66.67%
60.00%
NE
Run은 쉽게 시작하지만 완성이 상대적인 병목이 됨
다음 map의 가로축은 qualifying negative run을 시작하는 능력이다. 세로축은 이미 N2에 도달한 뒤 C를 완성하는 능력이다. Bubble 크기는 eligible sequence 수를 반영한다.
낮은 시작 + 낮은 완성
CRE stool과 VRE stool은 여러 gate에서 계속 어렵다.
낮은 시작 + 유리한 완성
CRE urine은 negative run이 드물게 시작되지만 N2 이후에는 자주 완성된다.
쉬운 시작 + 상대적으로 불리한 완성
MRPA sputum은 첫 음성만으로 진행을 과대평가할 수 있음을 보여준다.
유리한 시작 + 유리한 완성
VRE urine과 MRAB sputum이 가장 progressive한 영역에 위치한다.
어떤 층에서는 N1 진입 probability가 낮더라도 이미 N2에 도달한 뒤 C를 완성할 probability가 높을 수 있다. 두 확률은 서로 다른 source-state population을 조건으로 하므로 반드시 함께 움직일 이유가 없다.
Recurrence에는 명시적인 denominator가 필요하다
Recurrence endpoint
결과
의미
Transition-level C→P
29/69 = 42.03%
C에서 시작한 observed transition 중 positive next transition
Followed cleared sequences 중
21/39 = 53.85%
Post-C surveillance가 계속된 sequence에서 최소 한 번 recurrence
모든 cleared sequences 중
21/88 = 23.86%
Counted post-C transition이 없는 sequence도 포함함
Followed cleared patients 중
17/27 = 62.96%
Followed patient당 최소 하나의 recurrent sequence
첫 detected recurrence까지 시간
Median 11일; IQR 7–31일
검출된 recurrence는 흔히 이른 시기에 발생함
29/69
Transition-level recurrence
21/39
Followed-sequence recurrence
17/27
Followed-patient recurrence
11일
첫 recurrence까지 median
따라서 operational MM은 protocol을 설명하는 가장 강한 모델이지만, 배양 사이의 continuous biological state, measurement variation과 true burden change의 차이, pathogen–site 차이의 원인을 직접 규명하지는 못한다.
III. 2-regime switching-HMM: heterogeneous transition dynamics의 근거
R1과 R2가 의미하는 것
Switching-HMM은 네 개의 observed operational state를 그대로 유지한다. 여기에 observation step \(n\)에서 어떤 transition matrix를 사용할지 결정하는 hidden regime \(R_n\)을 추가한다.
R1과 R2는 hidden node이지만 biological burden state가 아니라 transition regime이다. Continuous-time model의 High Burden 및 Low Burden과 동일하지 않다.
두 transition phenotype은 현저히 다르다
Feature
R1
R2
해석
Estimated occupancy
68.36%
31.64%
Fitted observation interval의 대부분이 R1에 배정됨
P→N1
7.55%
54.34%
R2에서 negative run 시작이 훨씬 쉬움
N1→N2
28.51%
69.77%
R2에서 첫 음성이 훨씬 안정적임
N2→C
42.07%
79.30%
R2에서 완성 probability가 상당히 높음
C→C
16.46%
61.45%
R1에서는 operational C의 유지가 훨씬 불리함
Regime persistence
R1→R1 97.24%
R2→R2 99.67%
Regime switching은 가능하지만 observed transition당 드물게 발생함
전형적인 R1 journey
P → P → P → N1 → P
Persistence와 reset이 지배적이다.
전형적인 R2 journey
P → N1 → N2 → C
Forward progression이 지배적이다.
결과의 의미: 하나의 pooled transition matrix는 매우 다른 두 interval type을 평균낸다. 데이터는 predominant persistent/reset-prone mode와 더 작은 clearance-progressive mode로 설명될 때 더 잘 맞는다.
2-regime model은 MM보다 AICc를 약 90점, BIC를 약 39점 낮추고 patient-separated log loss를 약 4% 개선했다. 4-regime model은 한 번의 partition에서 CV loss가 조금 더 낮았지만, 2-regime model과의 상대적 predictive gap은 0.82%뿐이고 BIC는 약 165점 더 나빴다. 따라서 2개 regime이 fit, prediction, occupancy, separation 및 parsimony의 가장 방어적인 균형을 제공한다.
Regime이 증명하지 못하는 것
R1이 자동으로 CRE stool, 중증 환자, 항생제 노출 또는 하나의 고정 환자군을 뜻하지 않는다.
R2가 자동으로 biological clearance 또는 영구적으로 favorable한 patient class를 뜻하지 않는다.
Sequence는 R1과 R2 사이를 이동할 수 있으나 fitted regime은 매우 persistent하다.
Pathogen, site, treatment, device 및 host condition이 regime membership을 설명할 가능성은 있지만 해당 explanatory regression은 아직 완료되지 않았다.
지지되는 문장
지지되지 않는 과잉해석
두 종류의 transition-dynamics pattern이 지지된다.
두 종류의 생물학적 환자형이 발견되었다.
R1은 더 persistent하고 reset-prone하다.
R1은 하나의 pathogen, site 또는 treatment가 원인이다.
R2는 더 clearance-progressive하다.
R2는 biological eradication을 증명한다.
Regime은 시간에 따라 바뀔 수 있다.
각 sequence는 하나의 regime에 영구적으로 속한다.
따라서 switching-HMM은 transition behavior가 heterogeneous하다는 결론을 강화하지만, heterogeneity의 생물학적 원인을 규명하지는 못한다.
IV. Continuous-time latent process와 hidden semi-Markov 질문
가장 짧은 설명: snapshot 사이의 movie를 복원하는 model
4-state MM과 switching-HMM은 culture date에서 보이는 sequence를 분석한다. Continuous-time latent model은 그 visible point들을 연결했을 가능성이 있는 biological path를 질문한다.
Snapshot analysis
Day 0 positive
Day 7 negative
Day 28 positive
기록된 결과는 알 수 있다.
Continuous-time analysis
High Burden이 Low Burden으로 감소하고, 변동하고, Clearance에 접근했다가 다음 culture 전 rebound했을 가능성을 추정한다.
관찰되지 않은 path는 확률적으로 추론된다.
Day 0 Positive
관찰되지 않은 7일
Day 7 Negative
관찰되지 않은 21일
Day 28 Positive
관찰된 정보는 세 점뿐이다. Latent process가 day 0 직후 변했는지, day 7 직전에 변했는지, day 7과 day 28 사이에 여러 번 변했는지, 또는 전혀 변하지 않았는지를 직접 알 수 없다. 정확한 biological transition time은 interval-censored되어 있다.
가장 중요한 해석: 이 model은 가능한 trajectory의 probability distribution을 추정한다. 하나의 확실한 hidden movie나 하나의 정확한 transition date를 복원하는 것은 아니다.
Continuous-time, hidden, Markov, semi-Markov의 의미
Continuous-time
Latent transition은 다음 culture가 채취되는 순간에만 발생하는 것이 아니라 언제든 발생할 수 있다. 따라서 3일 간격과 30일 간격은 서로 다른 transition opportunity이다.
Hidden
실제 biological state는 직접 관찰되지 않는다. 선택된 날짜에서 positive 또는 negative culture emission만 관찰된다.
Markov
현재 latent state가 이후 transition rate를 결정한다. 그 state에 이미 얼마나 오래 머물렀는지는 rate를 직접 바꾸지 않는다.
Semi-Markov
한 latent state에서 벗어날 probability가 그 state에 이미 머문 시간에 따라 달라질 수 있다. Duration이 model의 일부가 된다.
일반 CT-HMM의 짧은 시간 동안 exit probability는 대략 다음과 같다.
\[
\Pr(\text{exit during }dt\mid X(t)=i)
\approx q_i\,dt.
\]
여기서 \(q_i\)는 해당 state에 이미 머문 시간에 직접 의존하지 않는다. Hidden semi-Markov model에서는 elapsed duration \(u\)가 hazard를 바꿀 수 있다.
\[
\Pr(\text{exit during }dt\mid X(t)=i,\text{ duration}=u)
\approx h_i(u)\,dt.
\]
Duration chart는 개념도이다. Duration-independent Markov rate와 duration-dependent semi-Markov rate의 차이를 설명하기 위한 그림이며, 현재 MDRO dataset에서 fitting된 curve가 아니다.
Markov와 semi-Markov는 같은 것이 아니다
Feature
Continuous-time HMM
Continuous-time hidden semi-Markov model
Latent state
Hidden
Hidden
Transition timing
Continuous
Continuous
State에 이미 머문 시간
Exit rate를 직접 바꾸지 않음
Exit probability를 직접 바꿀 수 있음
Dwell-time distribution
일반적으로 exponential
Non-exponential distribution 허용 가능
임상적 예
High Burden 5일째와 100일째의 instantaneous exit rate가 같음
오래 지속된 High Burden이 새로 진입한 High Burden보다 벗어나기 어려울 수 있음
Complexity
낮음
높음; duration에 대한 더 많은 정보가 필요함
현재 앱이 실제로 fitting한 model
중요한 용어 구분: 현재 primary result는 Three-State CT-HMM이다. 완전한 unrestricted continuous-time hidden semi-Markov model은 아니다. 별도의 duration-aware phase-type CT-HMM이 semi-Markov behavior의 constrained approximation으로 fitting되었다.
Model
포함 내용
현재 분석에서 역할
현재 상태
Three-State CT-HMM
정확한 culture interval을 이용한 High Burden, Low or Intermittent Burden, Sustained Clearance
각 latent state의 explicit하고 flexible한 dwell-time distribution
현재 implementation에서 fitting되지 않음
아직 미구현
H1
High · early
→
H2
High · established
→
L1
Low · early
→
L2
Low · established
→
C
Sustained clearance
L1 또는 L2는 H1 쪽으로 rebound할 수 있음
C는 L1 쪽으로 돌아갈 수 있음
Phase expansion은 state duration에 대한 일부 memory를 제공한다. H2에 있는 process는 구조상 H1에 있는 process보다 High Burden에 더 오래 머물렀다. 그러나 이는 practical semi-Markov approximation이며 unrestricted dwell-time curve를 직접 추정하는 것은 아니다.
현재 primary model은 three-state CT-HMM이다
High Burden
Culture-positive probability 98.88%
⇄
Low or Intermittent Burden
Culture-positive probability 83.57%
⇄
Sustained Clearance
Culture-positive probability 13.02%
Latent state
Positive emission
Negative emission
실제 해석
High Burden
98.88%
1.12%
거의 항상 positive로 보인다.
Low or Intermittent Burden
83.57%
16.43%
Latent burden이 남아 있어도 때때로 negative로 보일 수 있다.
Sustained Clearance
13.02%
86.98%
대부분 negative이지만 fitted state가 완전한 absorbing state도, 완벽하게 관찰되는 state도 아니다.
이들은 probabilistic latent state이다. Low or Intermittent Burden은 negative culture와 동일하지 않다. Fitted model은 이 state에서도 83.57%의 positive-emission probability를 부여한다. Sustained Clearance 역시 완전한 absorbing state가 아니다. Positive emission이 가능하며 model은 Low Burden으로의 return을 허용한다.
Model comparison은 three-state를 지지하지만 unrestricted semi-Markov model을 지지하지는 않는다
Prespecified 및 criterion-selected; flat likelihood ridge가 남음
Duration-Aware Phase-Type CT-HMM
12
2,145.026
2,213.495
0.475017
CV advantage는 작고 objective stability에는 미도달
Three-state model은 two-state model보다 AICc를 17.16점 개선한다. 두 모델의 BIC 차이는 0.04점뿐이므로 사실상 동률이다. Duration-aware model은 three-state model보다 CV log loss를 0.00123, 약 0.26% 개선하지만 BIC는 38.60점 나쁘다.
따라서 intermediate Low or Intermittent Burden state를 유지하는 해석은 타당하지만, 실제 생물학적 상태가 정확히 세 개라고 증명된 것은 아니다. 더욱 중요한 점은 duration-aware extension이 simpler three-state CT-HMM을 대체할 만큼 충분한 지지를 받지 못했다는 것이다.
직접적인 답변: dataset은 continuous-time latent modeling을 주의를 전제로 지지한다. 그러나 full hidden semi-Markov duration model이 반드시 필요하다는 더 강한 주장은 현재 지지하지 않는다.
시작, rebound, 완성 및 recurrence가 continuous-time rate로 바뀐다
Latent transition
100일당 rate
Single-edge expected waiting time
생물학적 해석
High → Low
1.0056
99.4일
High burden에서 감소가 시작됨
Low → High
0.6452
155.0일
High detectability 쪽으로 rebound
Low → Clearance
2.8338
35.3일
Sustained clearance로의 완성
Clearance → Low
2.8001
35.7일
Latent recurrence 또는 renewed intermittent detectability
High Burden
1.0056 / 100일initiation →← rebound 0.6452
Low or Intermittent Burden
2.8338 / 100일completion →← recurrence 2.8001
Sustained Clearance
가장 느린 principal step은 High→Low initiation이다. Low Burden에 들어간 뒤에는 completion rate가 rebound rate보다 높다.
Constant-rate competing-exit interpretation에서 Low Burden의 다음 exit가 Sustained Clearance로 향할 conditional probability는 다음과 같다.
이 수치는 직접 관찰된 patient probability가 아니라 model-derived calculation이다. 그러나 operational model과 동일한 단계별 논리를 강화한다. Lower-burden process에 진입하는 것과 sustained clearance를 완성하는 것은 서로 다른 사건이다.
Pathogen–site continuous-time domain map
다음 map은 가로축에 regularized High→Low initiation rate ratio, 세로축에 Low→Clearance completion rate ratio를 배치한다. 1보다 크면 pooled three-state model보다 빠른 rate이다. Bubble 크기는 included sequence 수를 대략 반영한다. Sparse cell은 principal map에서 제외하였다.
Stratum
Sequences
Initiation RR
Completion RR
Rebound RR
Recurrence RR
Mean High
Mean Clearance
CRE · Stool
60
0.880
0.695
2.209
1.265
54.48%
11.55%
CRE · Sputum
23
0.927
1.054
1.307
1.681
37.24%
19.59%
CRE · Urine
29
1.408
0.843
1.101
0.705
36.75%
24.34%
VRE · Stool
33
0.922
0.903
0.973
1.297
41.57%
17.87%
VRE · Urine
21
1.746
2.043
0.625
0.574
12.22%
50.60%
MRAB · Sputum
11
1.926
2.937
0.470
0.773
1.90%
58.21%
CRE stool과 VRE urine은 operational 및 continuous-time layer에서 가장 일관된 signal이다. CRE stool은 initiation과 completion이 느리고 rebound가 강하며 High Burden posterior가 높다. VRE urine은 initiation과 completion이 빠르고 rebound 및 recurrence rate ratio가 pooled model보다 낮으며 Sustained Clearance posterior가 높다.
CRE urine은 model을 분리해야 하는 이유를 잘 보여준다. Operational P→N1 probability는 절대적으로 낮지만 latent High→Low rate는 pooled rate보다 빠르다. 동시에 Low→Clearance rate는 pooled rate보다 낮다. Model-based interpretation으로는 high-burden state에서는 비교적 빨리 벗어나지만 stable negative sequence를 즉시 만들지 못하고 full completion 전 intermittently detectable state에 머무를 가능성을 생각할 수 있다.
현재 결과가 3번 model을 지지하는가
제안되는 결론
현재 근거
판정
서로 다른 culture interval을 동일한 한 step으로 처리해서는 안 된다.
79명, 198개 sequence, 2,445개 culture에 exact calendar interval이 사용되었다.
지지됨
Intermediate Low or Intermittent Burden state가 유용하다.
Three-state model은 two-state model보다 AICc를 17.16점 개선했고 patient-CV prediction도 조금 더 좋았다.
주의를 전제로 지지됨
Three-state structure가 모든 criterion에서 명확하게 우월하다.
Two-state와 three-state BIC 차이는 0.04점뿐이다.
결정적으로 확립되지 않음
Duration dependence 때문에 semi-Markov model이 반드시 필요하다.
Transition time은 culture 사이에서 interval-censored되어 있다.
지지되지 않음
Latent state가 실제 organism load를 직접 측정한다.
Quantitative microbiological gold standard 없이 binary culture emission에서 추정된 state이다.
지지되지 않음
3번 model의 판정: dataset은 continuous-time latent interpretation을 주의를 전제로 지지한다. Initiation과 completion을 분리하고 intermediate Low or Intermittent Burden state를 유지하는 해석은 유용하다. 그러나 duration 자체가 transition behavior를 바꾸는 full hidden semi-Markov model이 반드시 필요하다는 강한 주장은 현재 지지되지 않는다.
Likelihood surface가 비교적 flat하여 여러 인접 rate combination이 culture sequence를 비슷하게 설명할 수 있다.
Binary culture result만으로 quantitative organism burden을 직접 식별할 수 없다.
Emission probability는 sampling variation, assay behavior, 측정되지 않은 treatment 및 pathogen–site heterogeneity를 흡수할 수 있다.
Pathogen–site rate ratio는 pooled model 쪽으로 regularization되며 완전한 Bayesian hierarchy가 아니다.
Duration-aware candidate는 objective stability에 도달하지 못했으므로 exploratory로 유지해야 한다.
상대적으로 안정적
전체 likelihood level과 broad High–Low–Clearance interpretation
덜 안정적
Flat likelihood ridge 위에서 개별 transition rate의 정확한 위치
직접 관찰되지 않음
실제 quantitative organism burden, 정확한 transition time 및 unrestricted dwell-time distribution
따라서 continuous-time model은 직접 측정된 biological burden이 아니라 supported-with-caution latent process interpretation으로 표현하는 편이 타당하다.
V. Joint sampling–Death framework: 의미와 현재 지지되는 범위
가장 짧은 설명: movie와 camera를 함께 분석하는 framework
Culture record는 실제로 검체가 성공적으로 채취되었을 때만 만들어진다. Death는 이후의 culture opportunity를 영구적으로 종료한다. 따라서 observed culture history는 biological process와 observation process가 함께 만든 결과이다.
Biological process
검체 채취 여부와 무관하게 집락화 부담은 계속 변한다.
Sampling process
Culture timing은 surveillance schedule, specimen availability, clinical condition, device 또는 이전 결과에 영향을 받을 수 있다.
Observed result
Successful collection date에서만 positive 또는 negative가 관찰된다.
Death process
Death는 이후 culture를 채취하고 C를 확인할 모든 기회를 영구적으로 종료한다.
Death를 포함하는 목적: Death 전에 negative culture가 증가하는지만 확인하기 위한 분석이 아니다. Negative culture가 감소하는지, sustained-clearance probability가 낮아지는지, Death를 modeling한 뒤 biological transition rate가 달라지는지, Death가 C를 완성할 미래 기회를 제거하는지를 함께 확인한다.
세 종류의 observation distortion을 분리해야 한다
왜곡
예
일반 culture 분석이 잘못 해석될 수 있는 이유
Informative sampling
특정 latent state에서 culture가 더 자주 또는 덜 자주 채취됨
Sampling opportunity가 많은 state가 observed culture series에 과다 반영됨
Specimen unavailability
Sputum, urine 또는 stool specimen을 얻지 못함
Culture result가 생성되지 않는다. Missing result를 negative로 바꾸어서는 안 된다.
Death에 의한 terminal truncation
세 번째 qualifying negative를 채취하기 전에 환자가 사망함
Latent process가 개선되기 시작했더라도 이후 C에 도달하는 모습을 관찰할 수 없다.
Positive
→
First negative
→
검체 확보 불가
→
Death
이 sequence에는 두 번째 음성이나 세 번째 음성이 없다. 한 번의 음성, 한 번의 missing observation opportunity 및 terminal endpoint가 있을 뿐이다.
여기서 \(X\)는 관찰되지 않은 biological trajectory, \(N\)은 specimen-collection process, \(Y\)는 observed culture result, \(D\)는 Death process이다.
현재 앱이 실제로 fitting한 것
중요한 용어 구분: 현재 implementation은 아직 하나의 fully unified joint sampling–Death likelihood가 아니다. 여러 관련 분석을 연결한 coordinated framework이다.
Culture-only CT-HMM
+
Death-aware CT sensitivity
+
Observation-time audit
+
Reverse-time terminal models
+
C-versus-Death competing risk
현재 component
답하는 질문
단독으로 답하지 못하는 것
Death-aware CT-HMM sensitivity
Death를 포함하면 biological transition rate가 이동하는가?
Latent burden이 Death의 원인임을 증명하지 못함
Observation-time audit
Latent state별 fitted culture intensity가 다른가?
실제 clinical sampling mechanism을 확립하지 못함
Reverse-time terminal models
Death 근처에서 observed negativity와 latent-state probability가 변하는가?
Terminal illness의 인과 mechanism을 확인하지 못함
Competing-risk multi-state model
Confirmed C와 Death 중 어느 사건이 먼저 발생하는가?
C 이후 recurrence와 전체 latent burden trajectory를 modeling하지 않음
Death-aware sensitivity에는 patient-balanced composite likelihood가 사용된다. 따라서 여러 pathogen–site sequence를 가진 한 환자의 patient-level Death endpoint가 여러 번 완전한 가중치로 중복 계산되지 않는다.
Death가 만드는 세 가지 서로 다른 분석 질문
Terminal trajectory
Death에 가까워질수록 observed negative 또는 latent-clearance probability가 증가하는가, 감소하는가?
P, N1 또는 N2에서 confirmed C와 Death 중 어느 사건이 먼저 발생하는가?
이 질문들은 서로 연관되지만 대체할 수 없다. N2의 favorable C-before-Death probability는 Death 전에 negative culture가 증가한다는 증거가 아니다. Terminal negative decline만으로는 어느 operational 또는 latent gate가 악화되었는지 알 수 없다.
현재 각 component가 추가하는 정보
Component
질문
현재 결과
근거의 경계
Death-aware CT-HMM sensitivity
Death를 추가하면 latent biological rate가 달라지는가?
Shared Death intensity가 유지되었고 biological rate는 약 7%–13% 이동함
Death 근처에서 observed negative 또는 latent clearance가 변하는가?
Terminal negative surge는 없으며 sustained-clearance probability가 감소함
관찰 연구이며 수집된 culture를 조건으로 함
Competing-risk multi-state model
Confirmed C와 Death 중 어느 사건이 먼저 발생하는가?
C-before-Death probability가 P에서 N1, N2로 갈수록 상승함
First-event model이며 post-C recurrence는 제외됨
Death를 포함하면 rate는 중간 정도 변하지만 전체 architecture는 유지된다
Death structure
AICc
BIC
해석
Shared Death intensity
951.119
986.807
직접 비교 가능한 Death-structure pair에서 선호됨
State-specific Death intensities
952.952
997.500
추가 state-specific hazard가 정당화되지 않음
Biological edge
Death 추가 후 상대적 변화
해석
High→Low
−13.28%
Fitted initiation rate가 중간 정도 감소함
Low→High
−10.31%
Fitted rebound rate가 중간 정도 감소함
Low→Clearance
−7.13%
Completion estimate는 대체로 유사하게 유지됨
Clearance→Low
−7.04%
Recurrence-like dynamics가 대체로 유지됨
따라서 Death는 fitted colonization process와 전혀 무관하지 않다. Death를 포함하면 rate estimate가 이동한다. 그러나 기본 High–Low–Clearance architecture를 뒤집지는 않는다.
Shared-intensity result의 의미: Death가 colonization state와 무관하다는 뜻이 아니다. 현재 자료에서 각 latent biological state마다 별도의 Death intensity를 추정할 필요가 충분히 지지되지 않았다는 뜻이다.
이 sensitivity branch의 absolute culture-only 및 Death-aware rate는 제한된 endpoint-linked composite cohort를 사용한다. 따라서 full-cohort primary rate와의 직접 비교보다 relative change가 더 해석하기 적절하다.
Terminal change는 negative 감소와 sustained-clearance probability 감소를 보인다
Terminal comparison
Death cohort
Independent-censoring cohort
해석
Observed negative fraction
Terminal minus earlier: −8.53 pp; 95% interval −14.08 to −2.69
−2.05 pp; interval −15.07 to +9.53
Death 전 terminal negative surge가 지지되지 않음
Latent High Burden
+0.25 pp
−0.01 pp
Complete window에서 High Burden은 거의 변하지 않음
Latent Sustained Clearance
−3.23 pp
+3.19 pp
Clearance probability가 반대 방향으로 이동함
추론된 Low/Intermittent Burden 변화
약 +2.98 pp
약 −3.18 pp
세 posterior state 합을 이용한 arithmetic decomposition
Earlier sustained clearance 21.57%
→
Death-terminal sustained clearance 18.34%
+
Posterior mass는 주로 Low/Intermittent Burden 쪽으로 이동
Model-derived terminal pattern은 갑작스러운 High Burden surge보다 sustained clearance를 완성하거나 유지하지 못하는 현상과 더 잘 일치한다. 다만 이는 fitted CT-HMM을 조건으로 한 inference이며 terminal illness가 해당 변화를 일으켰다는 인과적 증거가 아니다.
Bayesian observed-culture model도 유사한 방향을 보였다. Death-terminal minus earlier negativity는 −2.24 percentage points였고 95% credible interval은 −8.19에서 +3.58이었으며 증가할 posterior probability는 22.85%뿐이었다. Death-aligned change는 censoring-aligned change보다도 상당히 불리했다.
Death 질문에 대한 직접적인 답변: Death를 고려했을 때 Death 전에 negative culture가 증가한다는 결과는 나타나지 않았다. 더 일관된 결과는 observed negative 감소와 sustained-clearance probability 감소였다.
Competing risk는 C를 완성할 남은 기회를 측정한다
Starting state
Eventual C before Death
Death before C
90-day C
90-day Death
90일 후에도 P/N1/N2
P
42.07%
57.93%
10.59%
23.41%
66.00%
N1
57.81%
42.19%
34.13%
18.11%
47.76%
N2
77.73%
22.27%
65.25%
10.04%
24.71%
P
긴 pathway가 남아 있음
→
N1
한 번의 negative가 누적됨
→
N2
qualifying negative가 한 번만 더 필요함
→
C-before-Death 상승
42.07% → 57.81% → 77.73%
N2에서 C-before-Death probability가 높은 이유는 qualifying negative가 한 번만 더 필요하기 때문이다. Negative culture가 인과적으로 Death를 예방한다는 뜻은 아니다. 이미 operational endpoint에 가까운 episode는 C까지 남은 pathway가 짧다는 뜻이다.
Observation-process 결과는 현재 unresolved이다
Publication-critical audit: exported narrative는 shared observation intensity가 선호되고 state-dependent culture timing에 대한 BIC support가 없다고 기술한다. 그러나 표시된 numerical criterion은 state-specific model에서 현저히 낮다. 이 내부 불일치가 해결되기 전에는 observation-process 결과를 과학적 결론에 사용해서는 안 된다.
Observation-time fit
AICc
BIC
수치상 해석
Shared intensity
7,342.259
7,380.883
더 높은 criterion value
State-specific intensities
6,448.795
6,497.031
AICc 893.46점, BIC 883.85점 낮음
Latent state
Fitted observation intensity/day
Expected days between cultures
Audit concern
High Burden
0.13086
7.64일
Approximately weekly surveillance와 양립 가능함
Low or Intermittent Burden
0.00106
941.02일
Scheduled surveillance design에서 비현실적임
Sustained Clearance
0.12088
8.27일
Approximately weekly surveillance와 양립 가능함
Low-Burden state의 극단적인 observation interval은 Poisson observation assumption에서 weak identification 또는 model misspecification 가능성을 시사한다. 941일 estimate를 literal clinical sampling interval로 받아들여서는 안 된다.
본 연구의 surveillance는 approximately weekly schedule에 기반한다. 따라서 Poisson observation-time model은 실제 clinician sampling mechanism을 그대로 나타내는 model이 아니라 sensitivity analysis이다.
Selection logic과 displayed criterion이 일치하도록 교정되기 전까지 가장 방어적인 결론은 informative culture timing의 영향은 unresolved라는 것이다.
현재 앱은 하나의 unified likelihood가 아니라 여러 linked component를 fitting한다.
아직 미구현
4번 model의 판정: 현재 결과는 Death를 competing process이자 observation-terminating process로 고려해야 한다는 점을 지지한다. 그러나 state-specific Death hazard는 지지되지 않았고 sampling process가 성공적으로 분리되었다고 판단할 수도 없다. 따라서 “joint sampling–Death model”은 현재 하나의 완성된 fitted model보다 지향하는 analytical framework를 나타내는 표현에 가깝다.
VI. 동일한 culture sequence를 네 model로 해석하기
Worked example
Day 0 Positive
→
Day 7 Positive
→
Day 14 Negative
→
Day 21 Negative
→
Day 28 Positive
→
Day 35 이후 culture 없음
4-state MM
P → P → N1 → N2 → P
Negative run이 N2에 도달했지만 C 전에 reset되었다.
Switching-HMM
각 operational transition에는 R1 또는 R2가 적용된다.
Progressive dynamics에서 reset-prone dynamics로 이동했을 수 있다.
Three-state CT-HMM
High → Low → High 쪽으로 possible rebound
Negative pair가 instantaneous biological clearance와 같을 필요는 없다.
Duration-aware approximation
High 또는 Low Burden에 새로 진입했는지 오래 머물렀는지에 따라 해석이 달라질 수 있다.
Duration dependence를 제한적으로 검정한다.
Death–sampling framework
이후 observation opportunity가 truncation되었을 수 있다.
No culture는 negative culture가 아니다.
Model
동일한 sequence의 해석
알 수 있는 것
알 수 없는 것
4-state MM
P→P→N1→N2→P
두 번의 음성이 누적되었지만 C 전에 run이 reset됨
배양 날짜 사이의 biological burden
2-regime switching-HMM
Transition pattern이 R1, R2 또는 두 regime 사이의 switch로 배정될 수 있음
해당 interval이 persistent 또는 progressive mode 중 어느 쪽과 유사한가
왜 해당 regime이 발생했는가
Three-state CT-HMM
Negative pair에서 posterior가 High→Low로 이동하고 이후 positive에서 High 쪽으로 rebound할 수 있음
배양 사이의 continuous probabilistic trajectory
직접적인 quantitative burden과 정확한 transition time
Duration-aware approximation
High 또는 Low Burden에 새로 진입했는지 오래 지속되었는지에 따라 transition interpretation이 달라질 수 있음
제한적인 duration-dependence test
Unrestricted dwell-time distribution
Death–sampling framework
이후 culture 부재가 follow-up termination, specimen availability, discharge 또는 Death와 관련될 수 있음
Future clearance opportunity가 얼마나 truncation되었는가
추후 실제로 관찰되었을 culture result
VII. 현재 dataset의 통합 해석
여러 model layer에서 일관되는 결론
질문
통합 답변
근거 model
Confidence boundary
Clearance의 가장 큰 병목은 어디인가?
시작 단계이다. P→N1은 16.78%뿐이고 latent High→Low initiation도 느리다.
Operational MM과 CT-HMM
Observed process에는 강한 근거가 있으나 biological mechanism은 latent함
Negative evidence는 점차 강해지는가?
그렇다. N1은 fragile하고 N2는 훨씬 유리하며 세 번째 음성은 여전히 의미가 있다.
Operational MM과 competing risk
Protocol-defined evidence sequence에는 강한 근거
하나의 transition matrix로 충분한가?
아니다. 두 hidden transition regime이 fit과 prediction을 상당히 개선한다.
Switching-HMM
Supported이나 regime의 원인은 미확인
Clearance는 즉각적인 binary switch인가?
그렇지 않을 가능성이 높다. Intermediate low/intermittent state가 model을 개선하고 isolated negative를 설명한다.
Three-state CT-HMM
Weak identification으로 supported with caution
Full semi-Markov model이 필요한가?
현재 근거에서는 아니다. Duration-aware candidate의 prediction gain은 작고 BIC penalty가 크다.
Duration-aware model comparison
Exploratory 수준
Confirmed local clearance는 durable한가?
반드시 그렇지 않다. Operational recurrence와 latent Clearance→Low dynamics가 모두 상당하다.
Operational recurrence와 CT-HMM
Follow-up dependent
Death 전에 negative가 급증하는가?
지지되지 않는다. Observed negativity는 대체로 낮아지고 latent sustained clearance도 감소한다.
Terminal, Bayesian 및 CT reverse-time 분석
Observational, non-causal
Death에 latent state-specific hazard가 필요한가?
현재 CT sensitivity에서는 아니다. Shared Death intensity가 선호되었다.
Death-aware CT-HMM
Endpoint-linked composite cohort에 한정됨
Sampling이 state-dependent한가?
Unresolved이다. 현재 narrative와 numerical criterion이 충돌한다.
Observation-process audit
Scientific interpretation 전 교정 필요
하나의 fully joint sampling–Death model이 fitting되었는가?
아니다. 현재 결과는 여러 linked analysis를 결합한다.
Implementation audit
향후 methodological extension
통합 signal
4-state MM
Switching-HMM
CT-HMM / semi-Markov
Death–sampling framework
Initiation이 어렵다
직접 지지
Regime-dependent 지지
High→Low가 느림
Secondary
Later negative evidence가 강하다
직접 지지
특히 R2에서 지지
Compatible
C-before-Death 상승
Clearance는 reversible하다
C→P 관찰
Regime별 C durability 차이
C→Low modeling
Post-C recurrence는 first-event model 밖
Duration dependence
Modeling하지 않음
Modeling하지 않음
Exploratory phase-type test
Primary가 아님
Terminal negative surge
이 질문용 model이 아님
이 질문용 model이 아님
Clearance posterior 감소
지지되는 surge 없음
Sampling-process distortion
Modeling하지 않음
Modeling하지 않음
Sensitivity audit만 수행
Audit discrepancy로 unresolved
Pathogen–site ecological synthesis
CRE stool은 가장 일관된 persistence signal이다. Negative run을 거의 시작하지 못하고 latent initiation 및 completion이 느리며 rebound가 강하고 High Burden에 머무는 비율이 가장 높다.
VRE urine은 흔한 stratum 중 가장 일관되게 유리하다. Operational initiation과 completion이 유리하고 latent initiation 및 completion이 빠르며 rebound와 recurrence rate ratio가 pooled model보다 낮다.
Urine overall은 initiation과 completion이 빠르고 rebound가 약하며 latent-clearance share가 높다.
MRAB 및 MRPA strata는 흔히 favorable conversion을 보이지만 여러 cell은 작거나 regularized되어 있어 정확한 biological constant로 해석해서는 안 된다.
CRE urine은 observed first negative와 latent High→Low transition이 같은 사건이 아님을 보여준다. High Burden에서는 상대적으로 빨리 벗어나더라도 sustained clearance 전 intermittently detectable state에 남을 수 있다.
CRE stool
느린 시작
느린 완성
강한 rebound
높은 posterior burden
VRE urine
빠른 시작
빠른 완성
약한 rebound
높은 posterior clearance
CRE urine
Operational N1은 드묾
Latent High→Low는 빠름
Full completion은 상대적으로 느림
MRPA sputum
Negative run은 쉽게 시작
완성이 상대적인 병목
VIII. 가장 방어적인 보고 hierarchy
권장되는 model 역할
Primary clinical interpretation
4-state operational MM
향후 model은 biological state, sampling, culture emission 및 Death를 함께 추정해야 한다.
현재 linked framework가 이미 하나의 completed joint likelihood이다.
최종 통합 해석
4-state MM이 가장 명확한 임상 메시지를 제공한다. Positive sequence의 대부분은 positive로 남고, 한 번의 negative는 fragile하며, 두 번의 negative는 강하지만 불완전한 evidence이고, 세 번째 negative는 실제 confirmation 역할을 유지한다. Post-clearance recurrence 때문에 local C를 permanent eradication으로 해석할 수 없다.
2-regime switching-HMM은 pooled transition matrix가 현저히 다른 두 dynamic mode를 가린다는 사실을 보여준다. Fitted interval의 대부분은 persistent, reset-prone regime에 속하고 더 작은 일부는 clearance-progressive regime에 속한다. 이들은 transition phenotype이며 검증된 biological class가 아니다.
Three-state CT-HMM은 가장 유용한 continuous interpretation을 추가한다. Principal latent bottleneck은 High Burden에서 벗어나는 과정이다. Low or Intermittent Burden에 도달하면 rebound보다 Sustained Clearance completion이 더 유리하지만, Low Burden에서도 culture positive가 흔하고 Sustained Clearance도 reversible하다. Biological information은 제공하지만 weakly identified되어 있으므로 operational result보다 secondary position에 두어야 한다.
Duration-aware phase-type model은 새로 진입한 latent state와 오래 지속된 latent state의 behavior가 다른지를 질문한다. 현재 predictive gain은 매우 작고 BIC는 상당히 나쁘며 optimization stability도 더 낮다. 따라서 현재 분석을 fully supported hidden semi-Markov model이라고 표현할 근거는 없다.
Death-aware 분석에서는 terminal follow-up이 artificial negative surge를 만들지 않았다. 더 일관된 pattern은 observed negativity와 sustained-clearance probability의 감소이며, High Burden의 큰 증가보다 low/intermittent burden 쪽으로의 posterior 이동이다. 그럼에도 negative evidence가 누적되면 C까지 남은 pathway가 짧아지므로 conditional C-before-Death probability는 높아진다.
Sampling component는 unresolved이다. 표시된 observation-process information criterion과 exported narrative가 충돌하고, fitted Low-state observation interval은 approximately weekly surveillance에서 임상적으로 타당하지 않다. Fully unified joint sampling–Death likelihood는 아직 fitting되지 않았다.
Observed rule
P → N1 → N2 → C
Hidden transition mode
R1 ↔ R2
Latent biology
High ↔ Low ↔ Clearance
Duration question
Early versus established phases
Terminal context
Sampling + Death + unresolved follow-up
가장 방어적인 종합 문장: 간헐적 MDRO culture는 단계적이고 heterogeneous한 clearance process를 보여준다. 관찰된 protocol은 4-state MM, transition heterogeneity는 2-regime switching-HMM, 관찰되지 않은 interval process는 three-state CT-HMM으로 가장 적절하게 설명된다. Full semi-Markov duration dependence는 현재 primary structure로 지지되지 않는다. Death는 confirmation opportunity를 truncation하며 terminal endpoint 근처에서 negativity와 sustained-clearance probability 감소와 연관된다. Informative sampling의 기여는 observation-process audit 교정 전까지 unresolved로 유지해야 한다.
IX. 처음 접하는 독자를 위한 최종 설명
3번 model을 한 문단으로 정리하면
3번 model은 biological state를 연속적으로 변하는 hidden process로 보고 culture result를 그 process의 간헐적인 observation으로 본다. 현재 dataset은 Three-State CT-HMM을 유용한 secondary interpretation으로 지지한다. 가장 큰 latent bottleneck은 High Burden에서 벗어나는 과정이며, intermediate Low or Intermittent Burden state는 한 번의 negative culture가 biological clearance와 같지 않을 수 있는 이유를 설명한다. 그러나 strict convergence 및 identifiability limitation이 남아 있고, duration 자체가 transition behavior를 바꾸어야 한다는 더 강한 semi-Markov 주장은 현재 model comparison에서 지지되지 않았다.
4번 model을 한 문단으로 정리하면
4번 model은 observed culture history가 specimen collection timing과 Death에 의한 observation termination으로 일부 만들어진 결과인지를 질문한다. Death와 관련된 component는 유용한 정보를 제공한다. Death는 fitted rate를 중간 정도 이동시키고, terminal negativity는 증가하지 않으며, Death 근처에서 sustained-clearance probability는 감소하고, Death는 clearance sequence가 완성되기 전에 C와 경쟁한다. 반면 sampling component는 current observation-process output의 내부 불일치와 비현실적인 Low-state sampling interval 때문에 unresolved이다. 따라서 fully unified joint sampling–Death model은 아직 확립되지 않았다.
가장 방어적인 두 줄 결론
Continuous-time 결론
Three-State CT-HMM interpretation은 주의를 전제로 지지된다. Full semi-Markov duration dependence는 exploratory하며 현재 primary structure로 지지되지 않는다.
Sampling–Death 결론
Death-aware sensitivity와 competing risk는 주의를 전제로 지지된다. Sampling-process separation은 unresolved이며 하나의 fully joint model은 아직 fitting되지 않았다.